
29.12C: Cystic Fibrosis
- Page ID
- 8374
Cystic fibrosis (CF) is an autosomal recessive disorder leading to respiratory congestion, multiple organ failure, and metabolic changes.
- Evaluate the factors involved in cystic fibrosis (CF)
Key Points
- Cystic fibrosis is characterized by abnormal transport of chloride and sodium across the epithelium, leading to thick, viscous secretions.
- CF is caused by a mutation in the gene coding for cystic fibrosis transmembrane conductance regulator (CFTR), which is required to regulate the components of sweat, digestive juices, and mucus.
- The hallmark symptoms of cystic fibrosis are salty tasting skin, poor growth, poor weight gain despite a normal food intake, accumulation of thick, sticky mucus, frequent chest infections, and coughing or shortness of breath.
Key Terms
- CFTR gene: The gene that encodes the CFTR protein is found on the human chromosome 7, on the long arm at position q31.2. from base pair 116,907,253 to base pair 117,095,955. CFTR orthologs have also been identified in all mammals for which complete genome data are available.
- cystic fibrosis: An inherited condition in which the exocrine glands produce abnormally viscous mucus, causing chronic respiratory and digestive problems.
- autosomal recessive: A mode of inheritance of genetic traits located on the allosomes (the sex determining chromosomes).
Cystic fibrosis (also known as CF or mucoviscidosis) is an autosomal recessive genetic disorder affecting most critically the lungs and also the pancreas, liver, and intestine. It is characterized by abnormal transport of chloride and sodium across an epithelium, leading to thick, viscous secretions. The name cystic fibrosis refers to the characteristic scarring (fibrosis) and cyst formation within the pancreas. Difficulty breathing is the most serious symptom and results from frequent lung infections that are treated with antibiotics and other medications. Other symptoms, including sinus infections, poor growth, and infertility affect other parts of the body. CF is caused by a mutation in the gene for the protein cystic fibrosis transmembrane conductance regulator (CFTR). The ΔF508 human genome mutation is characterized by the deletion of three base pairs in the CFTR nucleotide sequence, causing the loss of the amino acid phenylalanine located at position 508. This protein is required to regulate the components of sweat, digestive juices, and mucus. CFTR regulates the movement of chloride and sodium ions across epithelial membranes, such as the alveolar epithelia located in the lungs. Although most people without CF have two working copies of the CFTR gene, only one is needed to prevent cystic fibrosis due to the disorder’s recessive nature. CF develops when neither gene works normally (as a result of mutation) and therefore has autosomal recessive inheritance.
Cystic Fibrosis Respiratory Infections by Age: A graph depicting common bacteria found in the respiratory tracts of cystic fibrosis patients as a function of age.
Diagnosis and Symptoms
CF is most common among caucasians and can be diagnosed before birth by genetic testing, or by a sweat test in early childhood. Ultimately, lung transplantation is often necessary as CF worsens. The hallmark symptoms of cystic fibrosis are salty tasting skin, poor growth, poor weight gain despite a normal food intake, accumulation of thick, sticky mucus, frequent chest infections, and coughing or shortness of breath. Symptoms often appear in infancy and childhood, such as bowel obstruction due to meconium ileus in newborn babies.
Complications
Lung disease develops in CF as a result of clogging of the airways due to mucus build-up, decreased mucociliary clearance, and resulting inflammation. Inflammation and infection cause injury and structural changes to the lungs, leading to a variety of symptoms. In the early stages, incessant coughing, copious phlegm production, and decreased ability to exercise are common. Many of these symptoms occur when bacteria that normally inhabit the thick mucus grow out of control and cause pneumonia. In later stages, changes in the architecture of the lung, such as pathology in the major airways (bronchiectasis), further exacerbate difficulties in breathing.
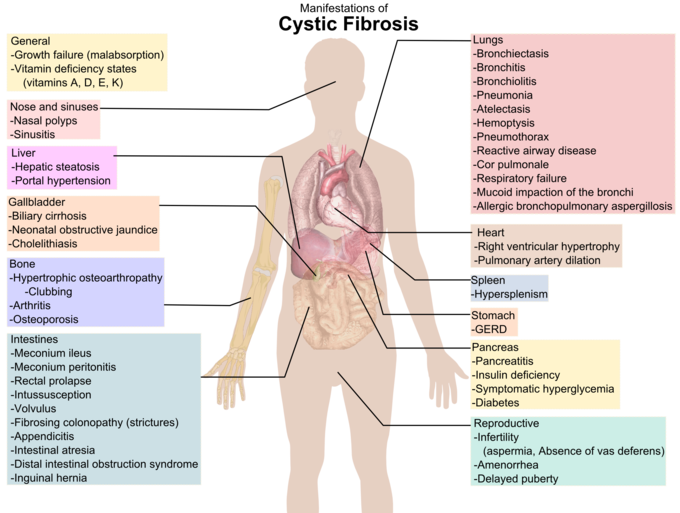
Clinical Manifestations of Cystic Fibrosis: A diagram showing clinical manifestations of cystic fibrosis.
Treatment
Many CF patients are on one or more antibiotics at all times, even when healthy, to prophylactically suppress infection. Antibiotics are absolutely necessary whenever pneumonia is suspected or there has been a noticeable decline in lung function, and are usually chosen based on the results of a sputum analysis and the patient’s past response. Several mechanical techniques are used to dislodge sputum and encourage its expectoration. In the hospital setting, chest physiotherapy (CPT) is utilized. As lung disease worsens, mechanical breathing support may become necessary. Bi-lateral lung transplantation often becomes necessary for individuals with cystic fibrosis as lung function and exercise tolerance declines.

A Deletion in the CFTR Gene is Responsible for CF: The ΔF508 human genome mutation is characterized by the deletion of three base pairs in the CFTR nucleotide sequence, causing the loss of the amino acid phenylalanine located at position 508.
Gene therapy has been explored as a potential cure for cystic fibrosis. Ideally, gene therapy attempts to place a normal copy of the CFTR gene into affected cells. Transferring the normal CFTR gene into the affected epithelium cells would result in the production of functional CFTR in all target cells, without adverse reactions or an inflammation response. Studies have shown that to prevent the lung manifestations of cystic fibrosis, only 5–10% the normal amount of CFTR gene expression is needed.
Finally, a number of small molecules that aim at compensating various mutations of the CFTR gene are under development. About 10% of CF cases result from a premature stop codon in the DNA, leading to early termination of protein synthesis and truncated proteins. One approach to combating a faulty receptor is to develop drugs that get the ribosome to overcome this premature stop codon and synthesize a full-length CFTR protein.