
7.2: Myocardial Disease - Primary
- Page ID
- 44018

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Five different groups of primary myocardial disease exist; which are defined as diseases of the myocardium with impaired cardiac function, also referred to as cardiomyopathies.
- Hypertrophic cardiomyopathy (HCM)
- Dilated cardiomyopathy (DCM)
- Restrictive cardiomyopathy (RCM)
- Arrythmic cardiomyopathy (ACM)
- Unclassified cardiomyopathy (UCM)
Hypertrophic cardiomyopathy
The essential characteristics of hypertrophic cardiomyopathy (HCM) are unexplained hypertrophy of the left ventricle in the absence of causal cardiac or systemic disorders. Distinctive features further comprise myocyte disarray, familial occurrence, and an association with sudden cardiac death (SCD).
Genetics
The identification of the genetic background of HCM resulted in the hypothesis that HCM is a disease of the sarcomere; the contractile unit of the cell. First, mutations were found in the cardiac B-myosin heavy chain gene, while later on other sarcomeric proteins were found to play a role in HCM (Table 7.3.1).
| Sarcomeric Genes Associated with HCM |
|---|
| α-Tropomyosin |
| Cardiac Troponin T |
| Troponin I |
| Myosin-binding Protein C |
| Regulatory Myosin Light Chain |
| Essential Myosin Light Chain |
| Cardiac Actin |
| Titin |
| Troponin C |
| α-Myosin Heavy Chain |
It was shown that macroscopic hypertrophy of the myocardium is not essential for neither diagnosis nor prognosis, as mutations for example in troponin T may lead to only minor or no hypertrophy, whereas it is associated with a high incidence of SCD. Nowadays HCM is considered a genetic disorder, inherited mainly autosomal dominant with an incomplete and age-related penetrance. Pathological findings may include myocardial hypertrophy, small-vessel disease and myocyte disarray with or without fibrosis. The prevalence of HCM in the adult population is approximately 1 in 500. But as in only 60% of HCM patients mutations in the abovementioned known sarcomeric genes are present, non-sarcomeric variants of HCM or mutations in regulatory genes, have gained interest. Left ventricular hypertrophy in young children is known most often not to be caused by sarcomeric HCM, but rather by inherited metabolic disorders and syndromes with extracardiac features. These diseases may cause to non-sarcomeric HCM as they include Fabry disease and Danon disease. Recognition and diagnosis of HCM may involve familial counselling, which also helps to find the presence of extracardiac features, and may influence treatment.
Epidemiology
The prevalence of the HCM phenotype was found to be approximately 0.2%, or 1 in 500, in several epidemiological studies. This frequency is notably higher than its occurrence in daily clinical practice. Hence, a large amount of patients remains undiagnosed, most probably without symptoms of inferences for their prognosis.
Pathophysiology
Left ventricular outflow tract obstruction (LVOTO)
The subdivision of HCM into obstructive and non-obstructive forms is clinically important, and is based on the presence or absence of a LV outflow tract gradient during rest and/or after provocation. The presence of such a gradient typically results in a loud apical systolic ejection murmur. The obstruction leads to an increase in intraventricular pressures, impairing LV function by increasing myocardial wall stress and oxygen demand. The obstruction is located either sub-aortic or mid-cavity, where the sub-aortic location is most common and is caused by systolic anterior motion (SAM) of the mitral valve and mid-systolic contact with the ventricular septum. SAM is thought to be facilitated by either an abnormal valvular apparatus loose enough to allow movement, or a hemodynamic force with an anterior component during systole. A drag-effect probably attributes to SAM, which refers to the force exerted by a fluid in the direction of the flow. Apart from its role in sub-aortic obstruction, SAM also results in concomitant mitral regurgitation. The sub-aortic gradient and associated LV pressure increase are pathophysiologically significant and may worsen outcome. LVOTO is an independent predictor for HCM-related death, progression of the disease in terms of New York Heart Association (NYHA) class III or IV, and death due to heart failure or stroke. A gradient threshold of 30 mmHg is prognostically important, but a further increase is not associated with further increased risk. Chronic outflow tract obstruction results in an increase in LV wall stress, myocardial ischemia and fibrosis, and justifies intervention in severely symptomatic patients when optimal medical management is insufficient.
HCM patients can be divided into hemodynamic subgroups based on the representative obstructive gradient:
- Obstructive - gradient under resting conditions equal to or greater than 30 mm Hg (2.7 m/s by Doppler)
- Provocable obstructive (latent) - gradient less than 30 mm Hg under resting conditions and equal to or greater than 30 mm Hg with provocation
- Nonobstructive - less than 30 mm Hg under both resting and provocated conditions.
LV outflow gradients are routinely measured noninvasively with continuous wave Doppler echocardiography. To define provocable gradients, treadmill or bicycle exercise testing is the preferred stress test, given that HCM-related symptoms are typically elicited with exertion. Pharmacological induction of stress by intravenous infusion of dobutamine is considered contra-indicated.
Diastolic Dysfunction
HCM is, in contrast to other cardiomyopathies, characterized by early occurrence of diastolic dysfunction (both active and passive phases), while systolic function is typically preserved, even before signs of hypertrophy. The passive relaxation during filling of the ventricle is hampered by increased chamber stiffness, increasing filling pressures and decreasing myocardial blood flow. Isovolumetric relaxation in early diastole is prolonged in HCM. Diastolic dysfunction may well lie at the basis of heart failure in non-obstructive HCM with preserved systolic function.
Ischemia
Myocardial hypertrophy disrupts the subtle equilibrium of blood flow throughout the myocardial layers. Patients with HCM show an increase in coronary blood flow velocity compared to healthy individuals reflecting an autoregulatory decrease in microvascular resistance to adapt to an increase in oxygen demand during resting conditions. The coronary reserve is therefore partly exhausted. Apart from the changes in the coronary microcirculation, systolic extravascular compression might play a role. Most importantly, HCM results in a progressive mismatch between muscle tissue and vascular growth, resulting in a high risk of myocardial ischemia especially for the subendocardial layers. The presence of myocardial ischemia is an important determinant of progression of the disease as it promotes scarring and remodelling of the ventricle.
Arrhythmia
Myocardial fibrosis associated with HCM is an important arrhythmogenic substrate. Functional consequences of HCM may therefore provide a trigger for ventricular arrhythmias, i.e. ischemia and LVOTO, resulting in non-sustained ventricular tachycardia in approximately 20% of patients. Other functional consequences as diastolic dysfunction, mitral regurgitation, as well as LVOTO are associated with atrial fibrillation (AF) which is observed in 20-25% of HCM patients.
Wall thinning and Cavity Dilation
Over time, thinning of the hypertrophic LV wall may occur in patients with severe LVH which may account for the lack of marked echocardiographic LVH in the elderly. While mechanisms of LV remodelling in HCM are still to be defined, cavity dilation and hampered systolic function occur in less than 5% of patients.
Clinical diagnosis
The cornerstone of HCM diagnosis is represented by echocardiography and electrocardiography. In the overall population, the diagnostic criterion of HCM is a maximal wall thickness greater or equal to 15 mm. However, virtually any wall thickness may be associated with the presence of an HCM mutant gene, including wall thickness well within the normal range. Therefore, for both echocardiography and electrocardiography, criteria have been established for the diagnosis of HCM in patients with high risk for HCM. Specificity of these criteria relies significantly on the a-priori chance of HCM, and is therefore only pertinent in first-degree relatives of index cases with confirmed HCM, whom therefore all have a 50% a-priori chance of carrying an HCM mutation. HCM is confirmed in a first-degree relative when either 1 major or 2 minor echocardiographic criteria (Table 7.3.2), or 1 minor echocardiographic and 2 minor electrocardiographic criteria (Table 7.3.3) are present.
Echocardiography
Two-dimensional echocardiography is the easiest diagnostic modality for detection of HCM, (Table 7.3.2) but cardiac magnetic resonance imaging (CMR) may be used when echocardiography is inconclusive, acoustic windows are insufficient, or when more detailed anatomic information is needed for clinical decision making. Echocardiographic characteristics include thickening of the left ventricular wall without cavity dilatation, and a normal or hyperdynamic left ventricle. Left ventricular outflow tract obstruction is not mandatory for the diagnosis of HCM. Moreover, as mentioned previously, although the diagnosis of HCM is based on a cut-off value for maximal wall thickness of 15 mm in the overall population, multiple HCM-linked mutations are associated with only minor LVH, but represent a high risk of sudden cardiac death.
| Major | Minor |
|---|---|
|
|
Electrocardiography
Electrocardiographic signs of HCM are typical as the increase in myocardial tissue increases the size of the QRS complexes. Therefore, a typical ECG characteristic of HCM is that it meets voltage criteria for LVH, and shows changes in repolarization (Table 7.3.3).
| Major | Minor |
|---|---|
|
|
Medical treatment
Asymptomatic HCM patients should only receive drugs when symptoms of diastolic dysfunction are present. Verapamil is the treatment of choice, improving diastolic filling and relaxation of the ventricle, decreasing diastolic filling pressures.
In symptomatic patients, first line medical treatment consists of a calcium-channel blocker or a betablocker. Verapamil is the first choice, but diltiazem may be used as an alternative. Second, beta-blockers may be used when symptoms prevail, and can be used solitarily or in combination with a calcium-channel blocker. In severely symptomatic patients, diuretics may be used, but with high caution as a small drop in ventricular filling pressure may reduce stroke volume and cardiac output dramatically in HCM patients. A combination can be made with either calcium-channel blockers or beta-blockers.
Patients presenting with ventricular tachyarrhythmia or supraventricular atrial fibrillation, amiodarone may be used, and may even improve symptoms and prognosis. Disopyramide has negative inotropic action and results in peripheral vasoconstriction and may improve symptoms when diastolic dysfunction is most prominent (i.e. with preserved ejection fraction).
Invasive treatment of obstructive HCM
In patients where maximal medical treatment does not control the symptoms, invasive debulking of the myocardial septum may be considered when a marked outflow gradient is present. Treatment options comprise percutaneous alcohol septal ablation, or surgical septal myectomy.
Surgical myectomy has shown excellent long-term result, but 15-20% of patients may suffer from ventricular remodelling and dilatation of the left ventricle. Since the introduction of alcohol ablation, surgical myectomy is reserved for patients with HCM with concomitant disease that independently warrants surgical correction, such as coronary artery bypass grafting of valve repairs, in whom surgical myectomy can be performed as part of the operation.
Septal ablation may be considered in patients with outflow tract gradients of more than 30 – 50 mmHg at rest or 60-100 mmHg after provocation. By injection of 1-3 mL of pure alcohol over 5 minutes into the first or second septal branch, a small myocardial infarction is created. Furthermore, the alcohol induces septal hypokinesis, adding to a reduction of the outflow tract gradient. The gradient may resolve immediately, but this process may take weeks to months. When the outflow tract obstruction persists, patients can be treated a second time.
Dual Chamber Pacing
In patients with medically refractory symptoms, whom are suboptimal candidates for invasive treatment, permanent dual chamber pacing may be considered. Pacing may alleviate symptoms by decreasing the outflow tract pressure gradient. However, maintaining a reduction in gradient requires pre-exitation of the right ventricular apex and distal septum, and complete ventricular caption. For optimal results, this should therefore be performed in highly experienced centers only.

Prognosis and outcome
In general, symptoms of HCM increase with age. Mortality rates have been reported to account between 2 and 3% per year. Most importantly, patients with HCM may be at high risk of sudden cardiac death, which may even be its initial presentation, in particular in asymptomatic or mildly symptomatic young patients. HCM is the most common cause of SCD in young people, including athletes. The pathophysiological basis for this predilection is unclarified, and although SCD is most frequent in young people less than 30 to 35 years old, an increased risk for SCD extends thereafter. Although HCM presentation and clinical manifestation is heterogeneous, and it has a relatively low prevalence, clinical markers as shown in Table 7.3.4 may identify patients at high risk for SCD. Patients at high risk of SCD are eligible candidates for ICD implantation.
| Major | Minor |
|---|---|
|
|
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) is a primary myocardial disease characterized by ventricular dilatation (one or both ventricles) and impaired myocardial contractility. The impairment of myocardial function cannot be explained by abnormal loading conditions alone, such as valve disease or systemic hypertension. The prevalence of DCM is approximately 36 per 100,000; in at least 50% of patients with DCM, its cause cannot be determined which is referred to as idiopathic DCM. DCM is a condition of which causes and presentations are highly heterogeneous. The diagnosis of idiopathic DCM should only be made after exclusion of the specific cardiomyopathies with a dilated phenotype.
Genetics
The genetic background of DCM is not as clear as in HCM. Although previously thought to be sporadic, genetic transmission is now thought to account for 30-40% of cases. Multiple genes have been identified that are linked with the occurrence of DCM. Genetic disease may account in part for the primary forms of DCM, but importantly, genetic predisposure may well lead to DCM in case of exposure to precipitating factors such as (emotional) stress, excessive alcohol use or stress upon the cardiovascular system; secondary DCM.
The expression of DCM in the familial form is frequently incomplete, and hence its prevalence is supposedly underestimated to a large extent. Even minor abnormalities may progress into overt DCM, and accurate clinical screening of (asymptomatic) relatives is therefore mandatory for early identification of familial DCM cases.
Pathophysiology
In general, a wide variety of factors can induce or contribute to the development of DCM including arterial hypertension, myocarditis, alcohol abuse or tachyarrhythmias. A subsequent increase in wall stress combined with activation of neurohumoral pathways induces complex cellular and molecular maladaptation, and programmed cell death finally leads to a decrease in the number of functioning cardiomyocytes. This process of cardiac remodelling itself results in systolic and/or diastolic dysfunction, leading to increased wall stress, and thereby creating a vicious circle of progressive systolic dysfunction (Figure 7.3.2).

The failing myocardium has several distinct factors promoting apoptosis of cardiomyocytes in vitro; cathecholamines, wall stress, angiotensin II, nitric oxide and inflammatory cytokines. Hence, medical management of DCM aims at antagonizing these pathways, reducing stress signalling in, and remodelling of the failing heart.
Clinical diagnosis
The most common initial manifestation of DCM is heart failure, in which clinical symptoms do not differ from heart failure of other causes. An important feature of the physical examination is a gallop rhythm of S3 and S4. S3 and S4 may fuse in tachycardic patients with new onset of heart failure. Special attention should focus upon excluding valvular heart disease as a cause, and excluding right-sided involvement.
Diagnostic testing in DCM focuses on identification of reversible causes, which focuses on the history (alcohol use) but also includes plasma biomarker evaluation, non-invasive imaging, electrocardiography, and exercise testing.
Echocardiography is an important diagnostic modality in DCM, as it can be used to assess both the size and shape of the LV, but also to determine LV function by assessing the LV ejection fraction (LVEF). Furthermore, valvular heart disease or pericardial abnormalities can be excluded. CMR evaluation may contribute to identification of specific cardiomyopathic conditions, especially when acoustic windows are suboptimal. The cardiac response to exertion is an established risk factor in DCM patients, which may be evaluated by cardiopulmonary exercise testing. Dilatation of the ventricular cavity results in myocyte stretch. In response, B-type natriuretic peptide is released, which is a well-known neurohormone that can be used to evaluate progression of DCM and to guide medical treatment. High plasma concentrations (twice the ULN) are strongly associated with increased long-term mortality.
Electrocardiography does not provide an accurate diagnostic mean in DCM, but can identify several characteristics associated with unfavourable prognosis, or identify factors contributing to DCM, and therefore is an important modality in the evaluation of DCM patients. Sinus tachycardia is frequently present, and non-specific ST-segment or T-wave changes as well as changes in P-wave morphology may well be present. AF is an important feature associated with high mortality; its control may contribute to improve cardiac performance. Furthermore, the presence of AF may indicate tachycardia-induced DCM. 24-hour Holter monitoring can reveal decreased heart rate variability or complex ventricular arrhythmias which are associated with a high risk for mortality. Finally, prolonged QTc intervals are associated with high mortality.
Management of DCM
Management of symptoms and progression of DCM accord to those described in the management of heart failure. Hence, also in DCM, diuretics and neurohumoral antagonists provide the basis for management of symptoms, and preventive ICD or pacemaker implantation is indicated in selected patients. Most importantly, surgical or percutaneous correction of underlying conditions facilitating progression of DCM, such as coronary artery disease, valvular heart disease or congenital abnormalities is warranted.
Specific dilated cardiomyopathies
It is important to note that there are several causes of secondary DCM. A foursome of these is of utmost importance to recognize early on, as accurate diagnosis influences the patients treatment strategy and chance for complete recovery: Tako-tsubo, Peripartum cardiomyopathy, Tachycardia-induced cardiomyopathy, Alcoholic cardiomyopathy (all described under 'Myocardial Disease - Secondary').
Cardiomyopathy in muscular dystrophy
Defined as primary disorders of skeletal and/or cardiac muscles of genetic etiology, muscular dystrophies were primarily described based upon the distribution and extent of skeletal muscle involvement. The involvement of the heart was commonly attributed to processes extrinsic to the heart, resulting in restrictive lung disease, subsequent pulmonary hypertension, and secondary myocardial dysfunction. Intrinsic dysfunction is increasingly recognized as an important etiology for myocardial function impairment in the presence of muscular dystrophy. Typical forms of dystrophy are based on deficiency of dystrophin, of which mutations have been described in X-linked DCM. Furthermore, histological changes were found in the myocardium similar to those in skeletal muscles, which suggest a common etiology, and moreover cardiac manifestations may be present even in the absence of myopathic symptoms.
Treatment of cardiac dysfunction is treated according to the nature of cardiac involvement. Conduction disorders may present which require pacing, and standard heart failure therapy may be instituted in case of ventricular dilatation and functional impairment. Ventricular tachyarrhythmias may be found in particular in myotonic dystrophia, and require the implantation of an internal cardiac defibrillator to prevent its associated sudden cardiac death.
Prognosis and outcome
DCM has a highly variable clinical course. Approximately half of DCM patients respond well to routine heart failure medication, and a minority of patients even shows an improving clinical course. Conversely, a subgroup can be identified with a highly unfavourable clinical course, not responsive to heart failure medication and rapidly progressing to inotropy- or LVAD-dependency. Overall, 5-year survival rates approximate 30%.
Restrictive cardiomyopathy
Restrictive cardiomyopathy is characterized by an increase in ventricular wall stiffness, impairing its diastolic function. Systolic function is usually preserved in early stages of the disease, but may deteriorate with progression of the disease. RCM is less frequent in the developed world than the previously described HCM and DCM, but is an important cause of death in Africa, India, South and Central America, and Asia due to the high endemic incidence of endomyocardial fibrosis. The spectrum of restrictive cardiomyopathies can be classified as shown in Table 7.3.5, according to its cause.
The most important specific causes of RCM will be discussed in detail below.
An important differentiation is that between RCM and constrictive pericarditis. Constrictive pericarditis is similarly characterized by impaired ventricular filling with preserved systolic function, but may be adequately treated by pericardiectomy, which makes this distinction of major clinical importance.
| Noninfiltrative | Infiltrative | Storage Disease | Endomyocardial |
|---|---|---|---|
|
|
|
|
Infiltrative cardiomyopathy
Amyloidosis
Amyloidosis is a disease that results from tissue deposition of fibrils that have a distinct secondary structure of a beta-pleated sheet configuration, leading to characteristic histological changes. Amyloid depositions can occur in almost any organ, but usually remains clinically undetected unless extensive depositions are present.
Types of amyloidosis
The most frequent types of amyloidosis are the AL (primary) and AA (secondary) types. AL amyloidosis is a plasma cell dyscrasia, which can occur solitarily or in association with multiple myeloma. AA amyloidosis can be considered a complication of chronic inflammatory disease states such as rheumatoid arthritis, in which the depositions consist of fragments of serum amyloid A, which is an acute phase reactant.
Hereditary amyloidosis has been increasingly recognized in the last decade, and results from mutations in the gene for thransthyretin. Some mutations are clinically limited to the myocardium. Its incidence increases with increasing age, with a predilection for men, but its prognosis is better than that of the AL type. Senile systemic amyloidosis results from deposition of normal wild-type transthyretin. This form of amyloidosis is clinically predominated by an infiltrative cardiomyopathy, but progression is slow and prognosis is better than of other acquired forms.
Cardiac amyloidosis
Cardiac amyloidosis is a progressive infiltrative cardiomyopathy. The primary form carries the highest cardiac involvement of approximately one third to half of patients, where deposits may be present even in the absence of clinical symptoms. Secondary amyloidosis is less frequently accompanied by cardiac infiltration, approximately 5% of cases, and is less likely associated with ventricular dysfunction due to a smaller size and more favourable location of the depositions. Familial amyloidosis is associated with clinical signs of cardiac involvement in a quarter of patients, typically presenting after the age of 35 with a distinct involvement of the cardiac conduction system. In senile amyloidosis, the extent of deposits may vary widely from solitarily atrial involvement up to extensive ventricular infiltration.
Clinical manifestations
Apart from the occurrence of cardiac disease in the presence of known AL amyloidosis or connective tissue disease or other chronic inflammatory disorders, cardiac amyloidosis should be considered in case of:
- Restrictive cardiomyopathy of unknown origin
- Left ventricular hypertrophy with a converse low-voltage ECG
- Congestive heart failure of unknown origin, not responding to contemporary medical management.
Clinical diagnosis
Diagnostic testing should include a 12-lead ECG, possibly with Holter monitoring, and routine echocardiography. Specific characteristics are a low-voltage 12-lead ECG with increase septal and posterior wall ventricular thickness.
Physical Examination
A physical examination may reveal an elevated jugular venous pressure, and signs of systemic edema. Auscultation frequently reveals an apical murmur due to mitral regurgitation, and a third heart sound, but the presence of a fourth heart sound may exclude amyloidosis, as atrial infiltration causes impaired atrial contraction.
Electrocardiography
Routine 12-lead ECG shows low voltage in the limb leads, and a pseudoinfarct pattern in approximately 50% of patients. Furthermore, conduction abnormalities occur frequently, as does atrial fibrillation.
Echocardiography
Thickening of the left ventricular wall with diastolic dysfunction are early echocardiografic features of the disease. In advancing disease, wall thickening increases, resulting in a restrictive cardiomyopathy. “Sparkling” myocardium is a distinct characteristic of cardiac amyloidosis, referring to an increased echogenicity of the myocardium. However, only a minority of patients has this pattern. Doppler evaluation shows a restrictive pattern with E dominance and a short deceleration time. Furthermore, intracardiac thrombus is common, which is associated with atrial fibrillation and left ventricular diastolic dysfunction.
The thickening of the ventricular wall caused by amyloidosis may be misinterpreted as hypertrophy on echocardiography. An important distinctive characteristic of amyloidosis is the voltage-to-mass ratio. Unlike normal hypertrophic myocardium, the increased ventricular mass in amyloidosis is associated with a decrease in electrocardiographic voltage.
Management
Few treatments for cardiac amyloidosis exist, and those available are dependent on the type of amyloidosis present. Hence, typing of the disease is pertinent. AL amyloidosis may be treated with chemotherapy using alkylating agents alone or in combination with bone marrow transplantation. Heart transplantation in combination with bone marrow transplantation after high-dose chemotherapy was shown to be result in approximately a third of treated patients surviving over 5 years, but as the great majority of patients with AL amyloidosis has severe non-cardiac amyloidosis, most patients are not suitable transplant candidates. Patients with other types of amyloidosis frequently have less affected hearts, and progression of the disease is slow. AA amyloidosis may respond to anti-inflammatory and immunosupressive drugs that reduce production of the acute-phase reactant protein. If heart failure is present, it is usually more prone to routine medical treatment to reduce symptoms. If needed, heart transplantation can be performed successfully. In patients where transthyretine is the amyloidogenic protein, liver transplantation may be curative as tranthyretine is produced in the liver, but the cardiac disease may progress regardless in some patients.
Overall, caution should be taken in prescribing digitalis, nifedipine, verapamil and ACE-inhibitors to cardiac amyloidosis patients. There is a high susceptibility to digitalis intoxication, nifedipine-induced hemodynamic deterioration, verapamil-induced left ventricular dysfunction, and ACE-inhibitor induced profound hypotension. If atrial fibrillation is present, or systolic ventricular function is severely impaired anticoagulation is indicated to prevent intracardiac thrombi. In selected patients with conduction disorders, pacemaker implantation may be considered. If ventricular function is severely impaired, ICD implantation may be considered. Nonetheless, prognosis of especially AL amyloidosis is poor.
Sarcoidosis
Sarcoidosis is a multi-system inflammatory condition characterized by the formation of non-caseating granulomas, most frequently affecting the lungs and lymphatic system. Myocardial involvement is seen in one quarter of cases only. Genetic factors are suggested as there was found to be an aggregation of cases within families.
Pathophysiology
Non-caseating granulomas may infiltrate the myocardium, leading to fibrotic scarring of the myocardium. Involvement of the myocardium is usually patchy, resulting in a relatively high likelihood of false-negative results from biopsy. Cardiac sarcoidosis must be differentiated from chronic active myocarditis and giant cell myocarditis.
Clinical Diagnosis
Patients may present with syncope, heart block or congestive heart failure. Sudden cardiac death may well be the initial manifestation of the disease due to malignant ventricular arrhythmias, but both atrial and ventricular arrhythmia is common at initial presentation. Symptoms of heart failure may result from direct myocardial involvement, but can also be due to extensive pulmonary fibrosis; cor pulmonale.
Physical examination may include signs of extracardiac sarcoidosis, a right sided third heart sound, and both S3 and S4, as well as murmurs of tricuspid regurgitation or mitral regurgitation.
Initial consideration of the diagnosis is often based on chest radiographs showing bilateral hilar lymfadenopathy. CMR is emerging as a highly sensitive and specific test for sarcoidosis, and nuclear imaging techniques may show regional perfusion defect due to the granulomatous inflammation on SPECT, or focal uptake on PET CT. electrocardiography is useful to assess the extent of conduction system involvement. Echocardiography shows left ventricular dilatation with hypokinesis, right ventricular enlargement and hypertrophy and possibly left ventricular aneurysm formation.
Minimal evaluation of a patient suspected of cardiac sarcoidosis consists of a 12-lead ECG, Holter monitoring and echocardiography.
Management
Early detection of the disease is critical for its clinical course. Immunosuppression using corticosteroids to halt the progression of inflammation is the treatment of choice in sarcoidosis, to which myocardial dysfunction, conduction disturbances, and arrhythmias may all respond. Most important is the differentiation of sarcoidosis from giant cell myocarditis, which is a more aggressive disorder requiring intensive medical and mechanical support and frequently necessitating heart transplantation. Pacemaker or ICD implantation is indicated in patients with conduction disorders or malignant arrhythmias, as medical treatment is usually ineffective in these cases.
Storage diseases
Hemochromatosis
Hemochromatosis is defined as a disorder of the iron metabolism, resulting in accumulation of iron in parenchymal tissues. In particular cardiac, liver, gonadal and pancreatic involvements are typical for hemochromatosis, in which the toxicity of redox-active iron results in organ dysfunction. Typically, hemochromatosis leads to a combination of heart failure, cirrhosis, impotence, diabetes and arthritis. Although several organ systems are usually involved, cardiac complications predominate the initial presentation, which are dependent on the site and amount of cardiac depositions. DCM is the typical phenotype of cardiac involvement, but a restrictive pattern may be present.
The most frequent, adult-onset, form of hemochromatosis was found arise from mutations in the HFE-gene, coding for a transmembrane protein involved in iron uptake in the liver and the intestine. Less frequently, mutations in the transferrine 2-encoding gene may result in hemochromatosis. The juvenile form of hemochromatosis results from mutations in the genes encoding hepcidin and hemojuvelin. The disease may also be acquired and result from ineffective erythropoiesis secondary to a defect in hemoglobin synthesis, chronic liver disease, chronic excessive oral or parenteral intake of iron, or from multiple blood transfusions.
Three stages of the natural history of adult-onset hemochromatosis can be differentiated. The first stage, the biochemical stage, is characterized by an iron overload which remains confined to the plasma compartment. Transferrin saturation is increased. The second phase, the deposition phase, is characterized by iron accumulation in parenchymal tissues, accommodated by an increase in serum ferritine levels. The third and final stage is that of organ dysfunction. Juvenile hemochromatosis is a more rapidly progressing disease, which leads to early organ dysfunction, around the age of 30, and is frequently characterized by premature death due to severe cardiac complications.
The invariably present symptoms of heart failure may frequently be accompanied by arrhythmias, especially ventricular extrasystoles, supraventricular tachycardia, and atrial fibrillation or flutter; either due to atrial iron depositions, or ventricular dysfunction resulting in increased ventricular pressure. Furthermore, conduction system involvement may lead to AV block or sick sinus syndrome.
Diagnostic features
Symptoms at initial presentation may vary. Echocardiography may show increased left ventricular wall thickness, ventricular dilatation, and ventricular dysfunction. CMR imaging represents a sensitive mean and may aid in early detection of the disease. Electrocardiographic characteristics include ST-segment and T-wave abnormalities as well as supraventricular arrhythmias, but occur as the disease advances. Biochemical testing reveals increased elevated transferrine saturation, increase plasma iron levels with low or normal iron binding capacity.
Management
Repeated phlebotomy is the cornerstone of hemochromatosis treatment, although chelating agents such as deferoxamine may be considered. Early detection of the disease is critical, as depletion of iron overload may result in complete reversal of symptoms at this stage. Evidence was found that a threshold exists beyond which iron depletion does not result in recovery of function. At end-stage disease, heart transplantation is a viable option with good survival rates. Importantly, screening of first degree relatives is important to ensure early detection of hereditary forms of hemochromatosis.
Fabry disease (angiokeratoma corporis diffusum universale)
Fabry disease is an X-linked inheritable deficiency of the lysosomal alpha-galactosidase A, resulting in an accumulation of glycosphingolipids in the lysosomes. There is a wide variety of know mutations, which all result in a different level of alpha-galactosidase inactivity, and hence, clinical manifestation may range from isolated myocardial disease to systemic involvement.
Patients may suffer from angina pectoris and myocardial infarction due to the accumulation of the aforementioned lipids in the endothelium of the coronary arteries, but the epicardial vessels show no abnormalities on angiography. Ventricular function is hampered due to thickening of the ventricular walls, which results in impaired diastolic compliance with a preserved systolic function, and may even precede myocardial hypertrophy. Other common features of the disorder include systemic hypertension, congestive heart failure, and mitral valve prolapse. The surface electrocardiogram may reveal a short P-R interval, atrioventricular block, and ST-segment and T wave abnormalities. Echocardiography may be inconclusive, but CMR imaging may differentiate Fabry disease from other infiltrative processes. Definite diagnosis is made on endomyocardial biopsy. Treatment of Fabry is safe and effective, and consists of enzyme-replacement therapy.
Gaucher Disease
Gaucher disease is an inheritable deficiency of beta-glucosidase, resulting in accumulation of cerebrosides. Cardiac involvement results in impaired cardiac function due to reduced ventricular compliance. Treatment of Gaucher disease consists of enzyme replacement therapy, or liver transplantation as a last resort. Response to treatment with respect to recovery of symptoms is heterogeneous.
Glycogen storage disease
Glycogen storage disease may result in cardiac involvement in case of type II, III, IV and V, but survival until adulthood is rare except for type III disease. Cardiac involvement is characterized by left ventricular hypertrophy, electrographically and echocardiographically, but cardiac symptoms are frequently absent.
Endomyocardial fibrosis
Endomyocardial fibrosis is an important cause of congestive heart failure in equatorial Africa. Prevalence up to 20% has been reported, mostly familial in children or young adults, although symptoms occurred only in a minority of detected cases. The disease predominantly occurs in black individuals, but may rarely present in white subjects.
The fibrous lesions hamper cardiac function by impairing the inflow of the ventricles, affecting the left or both ventricles most commonly. Solitaire right-sided involvement is less frequent, occurring in approximately 10% of cases. Symptoms are concordant with the ventricles involved, an atrial fibrillation and ascites are known factors associated with a poor prognosis. Clinical diagnosis is based upon clinical presentation, lab testing, and angiography. Left-sided myocardial biopsy is relatively contraindicated, as it may result in systemic emboli.
Endomyocardial fibrosis is a relentless disease, half of patients not making it beyond 2 years after detection, depending on the extent of symptoms at presentation. Medical management is only effective in the early stages of the disease, and is aimed at relief of symptoms. Surgical correction of the clinical consequences of the disease, i.e. valve replacement, improves hemodynamic characteristics, but mortality is high, and fibrosis may reoccur.
Hypereosinophilic syndrome: Löffler Endocarditis
The hypereosinophilic syndrome is a systemic disease, involving several organ systems. Cardiac involvement, Löffler endocarditis, is usually present when eosinophil counts are high for a longer period of time. The eosinophilia itself may occur from several different causes.
Histopathology shows eosinophilic myocarditis extending into the subendocardium, endocardial thickening, and inflammation of the small intramural coronary vessels.
Clinical features of the disease are weight loss, cough, fever, and a rash. Cardiac involvement may not induce clinical symptoms in the early stages of the disease, but as the disease progresses as much as 50% of patients develop cardiomegaly or congestive heart failure.
Cardiomegaly may be suspected on chest radiography, and the electrocardiogram shows non-specific abnormalities of the ST-segment and T-waves. Conduction defects may also occur. Echocardiography often shows a normal systolic function, and may show apical obliteration by thrombus. Obliteration of the apex with a preserved systolic function is a key characteristic of the disease on angiography. Endomyocardial biopsy can provide the diagnosis, but may be false-negative.
Routine care applies to these patients. Diuretics and neurohumoral blockade are appropriate, as is anticoagulation. Corticosteroids and cytotoxic drugs increase survival in patients with Löffler endocarditis, and interferon may be used as a last option in refractory patients. Surgical therapy may be considered as palliative treatment in the fibrotic phase of the disease.
Arrythmic cardiomyopathy: Arrhythmogenic Right Ventricular Cardiomyopathy
Arrhythmogenic Right Ventricular Cardiomyopathy, (ARVC, or ARVD: Arrhythmogenic Right Ventricular Disease, also AC Arrhythmogenic Cardiomyopathy) is characterized by fatty replacement and fibrosis of the heart. Most commonly, the right ventricle apex and outflow tract are involved. However, the left ventricle can be affected too. As a result of the fatty replacement and fibrosis, ventricular arrhythmias are common in this disease and can lead to palpitations, syncope and sudden death. At more advanced ages, right ventricular failure can occur.
ARVC is a progressive disease, and its incidence is estimated to be 1:3.000-1:10.000. The disease usually manifests at adolescence. Although the diagnosis is more often confirmed in athletes, physical activity is not thought to have a causal relationship with the disease. ARVC can occur in families; more than 9 different chromosomal defects have been described, most often with autosomal dominant inheritance. One unique form of ARVC, called Naxos disease (after the Greek island where it was first diagnosed), has an autosomal recessive pattern of inheritance.

Diagnosis
ARVC is a difficult diagnosis to make. Therefore, the European Society of Cardiology has created a list of diagnostic criteria for the diagnosis of ARVC, which were updated in 2009 (Tables 7.3.6 - 7.3.11). An online calculator can help in assessing the risk in an individual patient.
| I. Global or regional dysfunction and structural alterations* | ||||
|---|---|---|---|---|
| Major | Minor | |||
| By 2D Echo: | By MRI: | By RV Angiography: | By 2D Echo: | By MRI: |
|
|
|
|
|
| *Hypokinesis is not included in this or subsequent definitions of RV regional wall motion abnormalities for the proposed modified criteria. | ||||
| II. Tissue Characterization of Wall | |
|---|---|
| Major | Minor |
| Residual myocytes <60% by morphometric analysis (or <50% if estimated), with fibrous replacement of the RV free wall myocardium in =1 sample, with or without fatty replacement of tissue on endomyocardial biopsy | Residual myocytes 60% to 75% by morphometric analysis (or 50% to 65% if estimated), with fibrous replacement of the RV free wall myocardium in =1 sample, with or without fatty replacement of tissue on endomyocardial biopsy |
| III. Repolarization Abnormalities | |
|---|---|
| Major | Minor |
| Inverted T waves in right precordial leads (V1, V2, and V3) or beyond in individuals >14 years of age (in the absence of complete right bundle-branch block QRS =120 ms) |
Inverted T waves in leads V1 and V2 in individuals >14 years of age (in the absence of complete right bundle-branch block) or in V4, V5, or V6 Inverted T waves in leads V1, V2, V3, and V4 in individuals >14 years of age in the presence of complete right bundle-branch block |
| IV. Depolarization/Conduction Abnormalities | |
|---|---|
| Major | Minor |
|
|
| V. Arrhythmias | |
|---|---|
| Major | Minor |
|
|
| VI. Family History | |
|---|---|
| Major | Minor |
|
|
| †A pathogenic mutation is a DNA alteration associated with ARVC/D that alters or is expected to alter the encoded protein, is unobserved or rare in a large non–ARVC/D control population, and either alters or is predicted to alter the structure or function of the protein or has demonstrated linkage to the disease phenotype in a conclusive pedigree. E.g.: in TMEM43, DSP, PKP2, DSG2, DSC2, JUP. | |
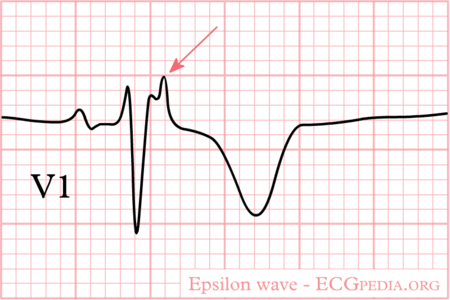
Treatment
Treatment focuses on avoiding complications.
- Medication:
- Anti-arrhythmics: Sotalol better than Amiodarone.
- ACE-inhibitors to prevent cardiac remodelling
- ICD implantation is recommended for the prevention of sudden cardiac death in patients with ARVC with documented sustained VT or VF who are receiving chronic optimal medical therapy.
- ICD implantation can be considered for the prevention of sudden cardiac death in patients with ARVC with extensive disease, including those with left ventricular involvement, 1 or more affected family member with SCD, or undiagnosed syncope when ventricular tachycardia or ventricular Fibrillation has not been excluded as the cause of syncope, who are receiving chronic optimal medical therapy, and who have reasonable expectation of survival with a good functional status for more than 1 year.
- Radiofrequency ablation can be useful as adjunctive therapy in management of patients with ARVC with recurrent ventricular tachycardia, despite optimal anti-arrhythmic drug therapy.
Unclassified Cardiomyopathy: Left ventricular non-compaction
Left ventricular non-compaction (LVNC) is characterized by distinct structural abnormalities:
A two-layered structure of the myocardium, with a thin compacted epicardial band and a thick non-compacted endomyocardial part, overall resulting in a thickened myocardium. LVNC is diagnosed by the ratio between the maximal non-compacted and compacted myocardial layer thickness measured during end-systole. In adults, a non-compacted/compacted thickness ratio of =2 confirms the diagnosis, whereas a ratio of =1.4 is considered diagnostic in children. The non-compacted layer is further characterized by prominent trabeculations, with intertrabecular spaces in direct connection with the left ventricular cavity. The non-compacted regions are predominantly found in the lateral, apical or inferior wall of the left ventricle. Where normal myocardium, originally consisting of a sponge-like structure of myocardial fibres, shows a pattern of basal to apical, and epicardial to endocardial compaction of the myocardial fibres during the 5th to 8th week of embryonal development, non-compaction supposedly originates from halting of this process due to a genetic defect.
Clinical Presentation and Diagnosis
Age of onset may be highly variable, with cyanosis, failure to thrive or dysmorphic features described in the neonatal period, to adult patients presenting with LV failure or ventricular arrhythmia. Possibly, sudden cardiac death entails one of the manifestations of LVNC, although evidence is only limited at this moment. Owing to technical advances in the field of echocardiography, predominantly an increase in image resolution, LVNC has only recently been recognized as an independent entity in the spectrum of cardiomyopathies, which supposedly has frequently been misdiagnosed as HCM in the past.
Two-dimensional echocardiography entails the cornerstone of LVNC diagnostics, enhanced by intravenous contrast agents if necessary. Magnetic resonance imaging may pose an option in case acoustic windows are insufficient. ECG abnormalities in patients with LVNC are mostly non-specific.
Considering the genetic background of LVNC, inheriting most commonly autosomal dominant, echocardiographic evaluation of family members of LVNC patients is pertinent.
Treatment and Prognosis
Treatment strategies are mainly extrapolated from other cardiomyopathies: standard heart failure therapy in case of LV dysfunction, beta-blockade and/or amiodarone in non-sustained VT in the absence of LV dysfunction, and ICD placement indicated in patients with LVEF <35%, sustained VT or recurrent unexplained syncope. Routine anticoagulation in not indicated, but anticoagulation may be instituted in case of ventricular dilatation or systolic LV function impairment.
The prognosis of LVNC has currently not yet been defined. One small report indicates a high mortality risk, owing to a high prevalence of sudden death, but community-based populations may well show a different prognosis as many patients with LVNC are non-symptomatic at the initial diagnosis.