
9.3: Arterial Vessel in Atherosclerosis
- Page ID
- 42773

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Three pathologic stages of atherogenesis
Atherogenesis can be divided into five key steps, which are 1) endothelial dysfunction, 2) formation of lipid layer or fatty streak within the intima, 3) migration of leukocytes and smooth muscle cells into the vessel wall, 4) foam cell formation and 5) degradation of extracellular matrix. Via these consecutive steps, an atherosclerotic plaque is formed. The formation of the plaque can also be divided into three major stages namely 1) the fatty streak, which represents the initiation 2) plaque progression, which represents adaption and 3) plaque disruption, which represents the clinical complication of atherosclerosis.

Initiation and formation of atherosclerotic plaque
The earliest visible signs of atherogenesis are the fatty streak and pre-existing lesions of adaptive intimal thickening. Fatty streak is a yellow discoloration on the surface of the artery lumen, which is flat or slightly elevated in the intima and contains accumulations of intracellular and extracellular lipid. At this stage of initiation, the fatty streak doesn’t protrude substantially into the artery wall nor impedes blood flow. This process is already visible in most people by the age of 20. At this stage, there are no symptoms and this lesion may even diminish over time. Initiation of fatty streak development is most likely caused by endothelial dysfunction, since it involves entry and modification of lipids within the subintima. This modified layer of lipids creates a proinflammatory environment and initiates the migration of leukocytes and formation of foam cells (Figure 9.3.2). Intimal thickening mainly contains smooth muscle cells and proteoglycan-collagen matrix with a few or no infiltrating inflammatory cells.

Endothelial dysfunction
Endothelial dysfunction is a primary event in atherogenesis, which can be caused by various agents, such as physical stress and chemical irritants. Endothelial dysfunction is also observed in other pathological conditions, which are often related to atherosclerosis such as hypercholesterolemia, diabetes, hypertension, heart failure, cigarette smoking and aging.
Factors Correlated with Endothelial Dysfunction
- Increased age
- Male sex
- Family history of coronary heart disease
- Tobacco smoking
- Elevated cholesterol
- Low HDL-cholesterol
- Diabetes mellitus
- Hypertension
- Obesity
- High fat consumption
Endothelial cells can display different reactions according to various levels of physical stress. There are two atheroprotective endothelial functions from physical stress. When endothelial cells are exposed to laminar flow, which display minimal physical stress, they secrete NO. NO functions as an anti-atherosclerotic substance through vasodilation, inhibition of platelet aggregation and anti-inflammatory effects. The second function is executed, when exposed to laminar flow by an expression of the antioxidant enzyme superoxide dismutase. This enzyme performs anti-atherosclerotic role by acting against reactive oxygen species, which are produced by chemical irritants or transient ischemia in the vessel.
Interventions that enhance endothelial function
- L-arginine
- Estrogen
- Antioxidants
- Quit smoking
- Reducing cholesterol
- Exercise
Unfortunately, these two atheroprotective endothelial functions can be impaired by several factors. The first factor is disturbed flow (low shear stress with rapid fluctuations), which is typically located at arterial branch points and bifurcations and can impair the protective functions. This is well illustrated by the difference in prevalence of atherosclerosis between branched arteries and bifurcated vessels. Bifurcation areas such as the common carotid and left coronary arteries are common deposition sites for atherosclerosis than arteries with few branches such as the internal mammary artery. Thus, many observations show that the distribution of atherosclerotic lesions is common in large vessels and they vary in location and frequency among different vascular beds. These findings encourage a belief that hemodynamic factors play an important role in atherogenesis. Furthermore, the fact that hypertension intensifies the severity of atherosclerotic lesions additionally supports this hypothesis.
Another major factor that can impair the atheroprotective endothelial function is chemical irritants such as cigarette smoking, abnormally high circulating lipid levels and high glucose level (diabetes mellitus). They can contribute to endothelial dysfunction and are all well- known risk factors for atherosclerosis. Exposure to chemical irritants promotes endothelial dysfunction by increasing endothelial production of reactive oxygen species, which alter the metabolic and synthetic functions of endothelial cells. As a result, the endothelium become inclined to exhibit proinflammatory processes, such as secreting inflammatory cytokines.
In conclusion, hemodynamic and chemical stressors contribute to disturbance of endothelial homeostasis and promote endothelial dysfunction. This results in impairment of permeability barrier function, secretion of inflammatory cytokines, stimulation of adhesion molecules on the cell surface that promote leukocyte recruitment, and altered antithrombotic properties and release of vasoactive molecules (Figure 9.3.3). Consequently, these effects establish the groundwork for further advancement of atherosclerosis.

Lipoprotein entry and modification
Disruption of the integrity of endothelial barrier due to endothelial dysfunction allows the passage of circulating lipoproteins (low-density lipoprotein, LDL) into the intima. By binding to proteoglycans, LDL particles start to accumulate. This accumulation is a critical process in atherogenesis since LDL may undergo chemical modifications while residing longer in the intima. It is needless to say that an elevated circulating LDL concentration strongly contributes to this accumulating process. Another major risk factor for this process is hypertension since it causes augmented vessel wall stress. Elevated vessel wall stress influences smooth muscle cells to synthesize proteoglycans in the intima, promoting LDL-binding with proteoglycans and therefore contributing to “trapping” of lipoproteins and lipid accumulation within the intima. At this point, macrophages adhere to dysfunctional endothelial cells and transmigrate into the intima. These macrophages are called ‘foam cells’ after they have taken up lipids.
As mentioned earlier, chemical modification occurs with LDL when chronic accumulation takes place inside the intima. There are several types of chemical modification that may occur. One is called oxidation and it results from the chemical reaction of reactive oxygen species and pro-oxidant enzymes produced by endothelial or smooth muscle cells, or macrophages penetrating the intima. This type of oxidative stress leads to cellular dysfunction and damage in endothelial cells and macrophages. Furthermore chronic hyperglycemia can stimulate glycation of LDL that may ultimately alter LDL into an antigenic and proinflammatory molecule. This explains why diabetes mellitus is a major risk factor for atherosclerosis. The biochemical modification of LDL into a proinflammatory molecule contributes to the inflammation process established by endothelial dysfunction. Furthermore, the oxidized LDL molecule induces tissue damage, which can initiate angiogenesis, forming new vasa vasorum in the plaque. It also induces leukocyte recruitment and foam cell formation in the fatty streak throughout the plaque development.
Leukocyte recruitment
Leukocyte recruitment to the arterial wall is another key step in atherogenesis, which is dependent on two important factors; expression of leukocyte adhesion molecules (LAM) on the endothelial wall and chemoattractant signals that direct diapedesis (intruding of molecules through the intact vessel wall). These two factors mainly direct monocytes to the atherosclerotic lesion. T lymphocytes that play a central role in the immune system reside within plaques at all stages of atherogenesis, mainly producing cytokines.
As mentioned earlier, modified LDL can maintain leukocyte recruitment by inducing LAM and chemokine expression. It can also stimulate endothelial and smooth muscle cells to produce proinflammatory cytokines. These proinflammatory cytokines can also induce LAM and chemoattractant cytokine expression, equivalent to the working of modified LDL. In conclusion, modified LDL can directly or indirectly promote leukocyte recruitment and atherogenesis.
Foam cell formation
When monocytes enter the intima, they differentiate into phagocytic macrophages. These phagocytic macrophages may become foam cells when they absorb lipoproteins. They don’t phagocyte LDL with a classic cell surface LDL-receptor, since it does not recognize modified LDL, but with a family of ‘scavenger’ receptors that do bind and internalize modified LDL. Uptake by scavenger receptors avoids negative feedback inhibition from the high cholesterol content unlike the classic LDL-receptors, and allows the macrophages to imbibe cholesterol-rich lipid that results into the formation of foam cells. This uptake seems to be beneficial at first sight, since it absorbs the inflammatory modified-LDL, however since these foam cells have impaired trafficking, they will be locally accumulated in the plaque and encourage the plaque progression by serving as a source of proinflammatory cytokines.
Plaque progression
The atherosclerotic plaque at this stage is called fibrous cap atheroma featuring two characteristics, which are lipid-rich necrotic core and encapsulation by a fibrous cap (Figure 9.3.4). The fibrous cap is an area between the vessel lumen and the core of the plaque, which contains dead foam cells, macrophages, smooth muscle cells, lymphocytes and extracellular matrix. A distinctive hallmark of this phase is necrosis with macrophage infiltration around a lipid pool and loss of proteoglycans or collagen. At this point, the deposition of free cholesterol is not easily visible and the plaque does not always cause luminal restriction of blood flow due to a compensatory outward remodeling of the plaque wall. This remodeling preserves the diameter of the vessel lumen and thus may evade detection by angiography. Continuous plaque growth at a later stage contains cellular debris, higher free cholesterol and results into complete depletion of extracellular matrix. From this stage, the fibrous cap atheroma may go through episodes of hemorrhage with or without calcification and even fibrous cap disruption. Progressive vessel narrowing may result in ischemia and can cause ischemic symptoms such as angina pectoris or intermittent claudication.
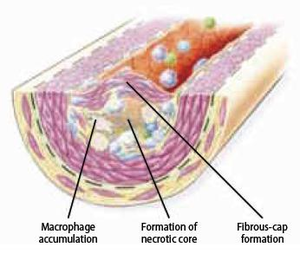
Smooth muscle cell migration
Smooth muscle cells play a central role at the phase of transition from fatty streak to plaque formation. During this phase, smooth muscle cells migrate from the media to the intima. After migration, smooth muscle cells proliferate within the intima and secrete extracellular matrix macromolecules. Additionally, foam cells, activated platelets and endothelium stimulate substances that induce the migration and accumulation of smooth muscle cells. For example, foam cells release platelet derived growth factor (PDGF), cytokines and growth factors that directly contribute to the migration and proliferation process, and they also activate smooth muscle cells and leukocytes to reinforce inflammation in the atherosclerotic lesion. Although plaque progression is traditionally known as a gradual and continuous process, recent evidence claims that this process can be strongly accentuated by bursts of smooth muscle replication. The observation of small ruptures within the plaque occurring without any clinical symptoms or signs supports this suggestion. These small ruptures expose tissue factor secreted by foam cells that stimulates coagulation and microthrombus formation in the lesion. Such microthrombi contain activated platelets that release additional factors such as PDGF and heparinase that can further stimulate local smooth muscle cell migration and proliferation. Heparinase stimulates smooth muscle cell migration and proliferation by degrading heparan sulfate, which normally counteracts this process.
Extracellular matrix metabolism
Metabolic processes in extracellular matrix play a central role in bridging the plaque progression to plaque rupture. Ultimately, this process weakens the fibrous cap, predisposing it to rupture. This process is influenced by the balance of matrix deposition synthesis by smooth muscle cells and degradation by matrix metalloproteinases (MMP), a class of proteolytic enzymes. For example, PDGF and TGF-β stimulate interstitial collagen production, while inflammatory cytokines such as IFN-γ inhibits collagen synthesis. TGF-β also induces formation of fibronectin and proteoglycans. It is an important regulator since it enhances the expression of protease inhibitors, leading to the inhibition of proteolytic enzymes that promote matrix degradation. On the other hand, inflammatory cytokines weaken the fibrous cap by stimulating local foam cells to secrete MMP that degrades collagen and elastin of the fibrous cap. Furthermore, the deeper parts of the thickened intima undergo necrosis due to poor nourishment.
Plaque rupture
Integrity of plaque
Chronic shifting of the balance towards extracellular matrix metabolism leads to serious consequences for the plaque integrity. As mentioned earlier, it accelerates inflammatory stimulation or activation of apoptosis pathways and therefore leads to death of smooth muscle and foam cells. Cell death leads to release of cellular contents, whereby more lipids and cellular debris is absorbed to the dynamic lipid core. Due to this process, the size of the lipid core grows and as a result alters biomechanical environment and hence the stability of the plaque. One example of this is a plaque border adjacent to the normal tissue, called shoulder region, which is the main location where the hemodynamic stress is focused. As the size and the protrusion of the plaque in the vessel increase, the hemodynamic stress will also increase around the shoulder region. Furthermore, local accumulation of foam cells and lymphocytes at this site makes the plaque more susceptible to rupture by accelerating degradation of extracellular matrix. However, although shoulder area is considered as the weakest point where the fibrous cap would mostly likely rupture, there have been autopsy studies that showed an equal number of ruptures occurring at the midportion of the fibrous cap. When the fibrous cap is very thick and contains small lipid core, the plaque is called stable and it may reinforce the narrowing of the artery, but on the other hand diminishes the susceptibility to rupture. Plaques with thinner fibrous caps are called vulnerable plaques. They are identified by a large necrotic core, rich with lipid, taking about 25% of the plaque area, and a thin fibrous cap of less than 65 µM thickness, which separates the necrotic core from the vessel lumen. Vulnerable plaque is infiltrated by a large amount of macrophages and a smaller amount of T-lymphocytes. It typically lacks smooth muscle cells due to apoptosis. This type of lesion causes less obstruction in the artery, but is more fragile and has higher susceptibility to rupture and trigger thrombosis than a thick fibrous cap. At this stage, plaque hemorrhage can occur due to rupture of vasa vasorum within a plaque. Vasa vasorum is a newly formed vascularization in the plaque due to tissue damage. Due to its fragility it may rupture easily, increasing the risk to form intraplaque hemorrhage. Intraplaque hemorrhage may lead to subsequent rupture of the fibrous cap (Figure 9.3.5) or occlusion of the vessel through intramural hematoma. Plaque calcification is another factor that contributes to plaque rupture. It usually occurs in areas of necrosis and elsewhere in the plaque and can eventually lead to higher rigidity of the vessel wall. Calcification is dependent on mineral deposition and resorption by osteoblast-like and osteoclast-like cells in the vessel wall. In conclusion, there are seven important factors associated with plaque ruptures; range of inflammation area, considerable size of lipid core, fibrous cap thinner than 65 µM, apoptosis leading to fewer smooth muscle cells, disrupted balance of proteolytic enzymes and their inhibitors, plaque calcification, and hemorrhage in the plaque. Although it remains difficult to foresee the clinical consequences, progression to a complicated plaque can lead to major cardiovascular disease, mostly affecting individuals in their 60s and 70s, although it may also occur among people at an earlier age.

Thrombogenic potential after rupture
When the fibrous cap is ruptured, the highly thrombogenic components of the necrotic core, including tissue factor, gets in direct contact with the circulating monocytes in the blood. It is believed that these circulating monocytes in the blood play a stronger role as a source of tissue factor than the necrotic core. Tissue factor stimulates platelet activation and thus can initiate and propagate thrombus. The thrombus formed at the rupture site is called white thrombus due to its grossly white appearance of rich platelet. At the proximal and distal ends near the site of white thrombosis there is another type of thrombus composed of layers of red blood cells and fibrin and is therefore called red thrombus. Thrombosis can be healed through several processes such as penetration of smooth muscle cells, neovascularization via vasa vasorum, proliferation of extracellular matrix, inflammation and re-endothelialization on the luminal surface. Thus clinically, ruptures can be silent and heal, without major clinical complications such as MI and stroke. For example, small non-occlusive thrombi may be reabsorbed into the plaque, continuing the process of smooth cell growth and fibrous deposition. The extent of how occlusive and transient the thrombus will be is largely dependent on the thrombogenic potential of the plaque.
The counter-balancing of coagulation and fibrinolysis also determines the probability of a major clinical event due to occlusive thrombosis. Inflammatory stimuli in the plaque environment incite smooth muscle cells, endothelial cells, and foam cells to release tissue factor that initiates the extrinsic coagulation pathway. Inflammatory stimuli also stimulate expression of antifibrinolytics such as plasminogen activator inhibitor-1 and consequently enhance thrombosis. As mentioned earlier, the activated endothelial cells also contribute to thrombosis and coagulation by depositing fibrin at the vascular wall. Thus, the inflammatory and dysfunctional condition of the plaque environment decreases the counterbalancing of coagulation and fibrinolysis, increasing the probability of major clinical complications of atherosclerosis.
The concept of ‘vulnerable plaque’ has developed into a new concept of ‘vulnerable patient’ as the concept of pathogenesis of atherosclerosis was linked to a person’s susceptibility to coagulation and thus vascular events, which can be influenced by many personal factors such as genetics (e.g. procoagulant prothombin gene mutation), coexisting condition (e.g. diabetes), and lifestyle factors (e.g. smoking, obesity).
