
11.8: Marfan Syndrome
- Page ID
- 42792

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Case report
Introduction
Marfan syndrome (MFS) is an autosomal dominant condition with a reported incidence of 1 in 3000 to 5000 individuals and is one of the most common inherited disorders of connective tissue. While most MFS patients have an affected parent, around 15 – 30 percent have a de novo mutation. MFS is associated with a broad range of clinical symptoms and associated disorders, ranging from classic ocular, cardiovascular, and musculoskeletal abnormalities to manifestations including involvement of the lung, skin, and central nervous system.
Progressive dilatation of the ascending aorta is one of the key features, which causes a high risk of sudden death due to aortic dissection or rupture in young Marfan patients. (Figures 11.8.1 & 11.8.2)
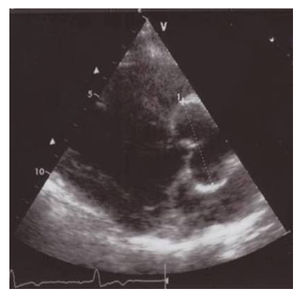
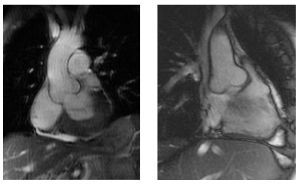
The underlying genetic defect is localised in the fibrillin gene on chromosome 15 (FBN1) in which recently around 600 different mutations are found. However in about 10% of MFS patients there is no mutation identified in the FBN1 gene, furthermore FBN1 mutations also occur across a wide range of milder phenotypes that overlap the classic Marfan phenotype. Therefore it is not possible to diagnose MFS solely with genetic information.
Criteria for diagnosis of Marfan syndrome
The 2010 revised Ghent criteria puts greater weight on aortic root aneurysm/dissection and ectopia lentis as the cardinal clinical features of MFS and on testing for mutations in FBN1 and other relevant genes.
In the absence of family history of MFS, the presence of one of any of the following criteria is diagnostic for MFS:
- Aortic criterion (aortic diameter Z≥2 or aortic root dissection) and ectopia lentis*
- Aortic criterion (aortic diameter Z≥2 or aortic root dissection) and a causal FBN1 mutation
- Aortic criterion (aortic diameter Z≥2 or aortic root dissection) and a systemic score ≥7
- Ectopia lentis and a causal FBN1 mutation as defined above that has been identified in an individual with aortic aneurysm
In the presence of family history of MFS (as defined by the above criteria), the presence of one of any of the following criteria is diagnostic for MFS:
- Ectopia lentis
- Systemic score ≥7 points*
- Aortic criterion (aortic diameter Z≥2 above 20 years old, Z≥3 below 20 years, or aortic root dissection)*
For criteria with an asterisk (*), the diagnosis of MFS can be made only in the absence of discriminating features of Shprintzen-Goldberg syndrome (SGS), Loeys-Dietz syndrome (LDS), or vascular Ehlers-Danlos syndrome (vEDS) and after TGFBR1/2, collagen biochemistry, or COL3A1 testing if indicated.
The revised Ghent nosology includes the following scoring system for systemic features:
- Wrist AND thumb sign: 3 points (wrist OR thumb sign: 1 point)
- Pectus carinatum deformity: 2 (pectus excavatum or chest asymmetry: 1 point)
- Hindfoot deformity: 2 points (plain pes planus:1 point)
- Pneumothorax: 2 points
- Dural ectasia: 2 points
- Protrusio acetabuli: 2 points
- Reduced upper segment/lower segment ratio (US/LS) AND increased arm span/height AND no severe scoliosis: 1 point
- Scoliosis or thoracolumbar kyphosis: 1 point
- Reduced elbow extension (≤170 degrees with full extension): 1 point
- Facial features (at least 3 of the following 5 features: dolichocephaly [reduced cephalic index or head width/length ratio], enophthalmos, downslanting palpebral fissures, malar hypoplasia, retrognathia): 1 point
- Skin striae: 1 point
- Myopia >3 diopters: 1 point
- Mitral valve prolapse (all types): 1 point
A systemic score ≥7 indicates systemic involvement.
Pathophysiology
Aortic root disease, leading to aneurysmal dilatation, aortic regurgitation, and dissection, is the main cause of morbidity and mortality in the MFS. Dilatation of the aorta is found in approximately 50 percent of children with MFS and progresses with time. Approximately 60 to 80 percent of adult patients with MFS have dilatation of the aortic root (with normal range adjusted for patient body surface area and age) by echocardiography, often accompanied by aortic regurgitation. Dilatation may also involve other segments of the thoracic aorta, the abdominal aorta, the root of the pulmonary artery or even the carotid and intracranial arteries. Untreated MFS is frequently associated with aortic dissection, which begins just above the coronary ostia and extends the entire length of the aorta; it is a type I dissection in the DeBakey classification or a type A in the Dailey scheme. Approximately 10 percent of dissections begin distal to the left subclavian (type III or type B) but dissection is rarely limited to just the abdominal aorta. Many patients with MFS and aortic dissection have a family history of dissection.
Mitral valve prolapse (MVP) is frequently identified in patients with MFS. However, only one point in the systemic score is assigned for MVP since it is a nonspecific feature and most patients with mitral valve prolapse do not have MFS. The frequency of MVP in MFS increases with age and is greater in women. Tricuspid valve prolapse may also occur.
Treatment
Beta blockers decrease myocardial contractility and may also improve the elastic properties of the aorta, particularly in patients with an aortic root diameter <40 mm thereby decreasing the risk of aortic dissection and delaying the aortic dilatation. Prophylactic treatment with beta blockers is considered the standard of care in adults with MFS. Furthermore patients with MFS are advised to avoid any contact sports, exercise at maximal capacity, and isometric activities.
The exact aortic root diameter at which elective surgery should be performed is uncertain. The current guidelines recommend elective operation for patients with MFS at an external diameter of ≥50 mm to avoid acute dissection or rupture. Indications for repair at an external diameter less than 50 mm include rapid growth (>2 mm/y), family history of aortic dissection at a diameter less than 50 mm, desire of pregnancy or presence of progressive aortic or mitral valve regurgitation. However one must take into account that a predicted aortic root diameter varies with body size and age and may be smaller in women. Smaller patients have dissection at a smaller aortic root size and 15 percent of patients with MFS have dissection at a diameter less than 50 mm.
The classic aortic root surgery is the Bentall procedure in which the ascending aorta is replaced, together with the aortic valve, by a graft with prosthetic valve. In this procedure the coronary arteries need to be reimplanted in the aortic graft. In patients with anatomically normal valves, in whom the insufficiency is due to the dilated annulus or dissection, valve-sparing operations with root replacement by a Dacron prosthesis and with reimplantation of the coronary arteries into the prosthesis (David’s procedure) or remodelling of the aortic root (Yacoub’s procedure) have now become the preferred surgical procedures. Aortic regurgitation is, however, a common complication, requiring reoperation in 20% of patients after 10 years.
Outcome
The reported operative mortality of the Bentall procedure is 1.5% for elective and 11.7% for emergency operations. Five- and 10-year survival rates of 84 and 75%, respectively, have been reported. This relatively limited prognosis is due to late sequelae associated with MFS; 75% require reoperations, 10% mitral valve surgery, 1% develop endocarditis and 1% a CVA. Marfan syndrome has also been associated with a considerably higher risk of re-dissection and recurrent aneurysm than other aetiologies of aortic disease. Long-term results of valvesparing aortic root replacement in Marfan syndrome are still unknown.