
1.2: Introduction to Pharmacology
- Page ID
- 10623

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A drug is a chemical agent which can affect living processes. For purposes of this course we will mainly be talking about small molecules which affect cellular processes. Most of these are Xenobiotics (Gr. xenos - stranger) chemicals that are not synthesized by the body, but introduced into it from outside. There is inevitably a certain amount of ambiguity in this definition: Is oxygen or water a drug? How about Vitamin C in a glass of orange juice? How about an injection of Vitamin C to treat scurvy?
Pharmacology (Gr. pharmakon - a drug or poison, logos - word or discourse) is the science dealing with actions of drugs on the body (pharmacodynamics) and the fate of drugs in the body (pharmacokinetics). It overlaps with pharmacy, the science of preparation of drugs; much of it deals with therapeutics, the treatment of disease (by whatever means). Toxicology is the branch of pharmacology dealing with the "undesirable" effects of drugs on biological processes (in the case of a nerve gas the bad effect may be a desired one).
In order for a drug to work, it must enter the body and somehow be distributed in such a way that it gets to its site of action. In most cases the site of action is a macromolecular "receptor" located in the target tissue. Most drug effects are temporary, because the body has systems for drug detoxification and elimination. We will consider these issues broadly for now and go into more depth in individual lectures. As you read, refer to the figure below:
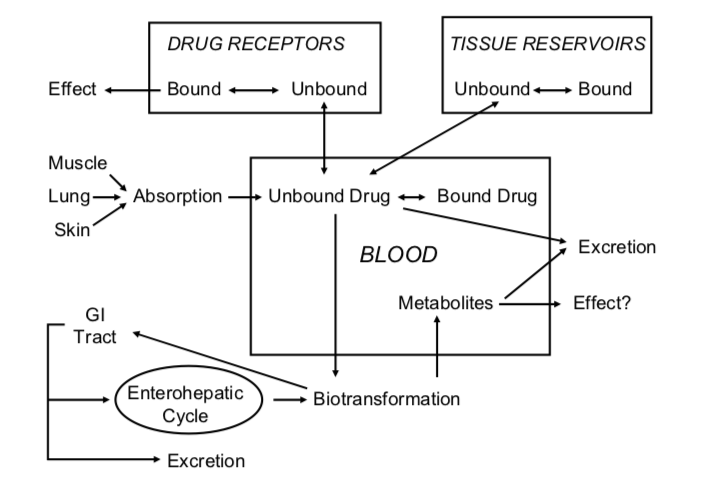
Overview of Pharmacokinetics - "What the body does to the drug"
- The drug may enter the body in a variety of ways: as an oral liquid, pill, or capsule; as an inhaled vapor or aerosol; absorbed through intact skin or a mucous membrane; injected into muscle, subcutaneous tissue, spinal fluid, or directly into the bloodstream. As we shall see, the physical properties of the drug and the specific way it is prepared greatly influence the speed of absorption.
- If the drug is given orally and swallowed, it must be absorbed from the GI tract into the portal circulation. If it is absorbed from the skin, mouth, lungs or muscle it will go directly into the systemic circulation. If drug is injected directly into the bloodstream (e.g., intravenous injection), 100% of it is available for distribution to tissues. This is not usually the case for other modes of administration. For example, drug which is absorbed via the portal circulation must first pass through the liver which is the primary site of drug metabolism (biotransformation). Some of the drug may therefore be metabolized before it ever reaches the systemic blood. In this case,"first-pass" metabolism reduces the bioavailability to less than 100%.
- Once the drug is in the bloodstream a portion of it may exist as free drug, dissolved in plasma water. Some drug will be reversibly taken up by red cells and some will be reversibly bound to plasma proteins. For many drugs, the bound forms can account for 95-98% of the total. This is important because it is the free drug which traverses cell membranes and produces the effect. It is also important because protein-bound drug can act as a reservoir which releases drug slowly and thus prolongs its action.
- The unbound drug may then follow its concentration gradient and distribute into peripheral tissues. In some cases, the tissue contains the target site and in others the tissue is not affected by the drug. Sites of non-specific binding act as further reservoirs for the drug. This total volume of distribution determines the equilibrium concentration of drug after a specified dose.
- Tissue-bound drug eventually reenters the bloodstream where it perfuses the liver and kidneys. The liver metabolizes most drugs into inactive or less active compounds which are more readily excreted. These metabolites and some of the parent compound may be excreted in the bile and eventually may pass out of the body in the feces. Alternatively, some of the drug may be reabsorbed again, farther down the GI tract (the so-called enterohepatic cycle). Any biotransformed drug which is not excreted in bile passes back into the systemic circulation.
- Parent drug and metabolites in the bloodstream may then be excreted: most are filtered by the kidney, where a portion undergoes reabsorption, and the remainder is excreted in the urine. Some drugs are actively secreted into the renal tubule. Another route of excretion is the lung: Drugs like alcohol and the anesthetic gases are eliminated by this route. Smaller amounts of drug are eliminated in the sweat, tears and breast milk.
- Biotransformation may sometimes produce metabolites with a great deal of activity. Occasionally, we administer a parent drug which is inactive (a pro-drug) and only the metabolite has activity. [How might this be useful?]
Overview of Pharmacodynamics - "What the drug does to the body"
As stated above, the majority of drugs bind to specific receptors on the surface or interior of cells, but there are many other cellular components and non-specific sites which can serve as sites of drug action.
- Water can be a target. Osmotic diuretics like mannitol are not reabsorbed by the kidney, and the osmotic load they create in the renal tubule obligates the loss of water. Laxatives like magnesium sulfate work in the intestine by the same principle.
- Hydrogen ions can be targets. Ammonium chloride is sometimes used to acidify the urine. When it is taken orally, the liver metabolizes ammonium ion to urea, while the chloride is excreted in the urine. The loss of Cl- obligates the loss of H+ in the urine, thus the pH is lowered.
- Metal ions can be targets. Chelating agents like EDTA may be used to bind divalent cations like Pb++. Metal ions are most frequently drug targets in cases of poisoning.
- Enzymes are targets of many therapeutically useful drugs. Drugs may inhibit enzymes by competitive, non-competitive, or irreversible blockade at a substrate or cofactor binding site. Digitalis glycosides increase myocardial contractility by inhibiting the membrane enzyme, Na+-K+ - ATPase. Antimicrobial and antineoplastic drugs commonly work by inhibiting enzymes which are critical to the functioning of the cell. In order to be effective, these drugs must have at least someselective toxicity toward bacterial or tumor cells. This usually means that there is a unique metabolic pathway in these cells or some difference in enzyme selectivity for a common metabolic pathway. An example of this is the inhibition of folate synthesis by sulfonamides. These drugs are effective antibacterial agents because the bacteria depend upon folate synthesis, while the host doesn't. This example will be covered in detail in one of our case discussions.
- Nucleic acids are targets for antimetabolites and some antibiotics. In the case of 5- fluorouracil, the compound acts as a counterfeit substitute for uracil and becomes incorporated into a faulty mRNA. Antisense oligonucleotides are another very specific way to interfere with a restricted part of the genome.
- Some drugs, like general anesthetics, appear to act by non-specific binding to a macromolecular receptor target. These drugs are thought to alter the function of membrane proteins, in part, by disordering the structure of the surrounding lipid membranes. Their lack of specificity is reflected in very low chemical structural requirements. The general anesthetics include compounds as chemically diverse as nitrogen, xenon, halogenated ethers, and steroids. They exhibit very little stereoselectivity, that is, there are not marked differences in anesthetic activity between enantiomers.
- Finally, we have the drugs which act by binding to specific receptors. As you will see in lectures 2 and 6, these drugs have both high structural specificity and stereoselectivity, i.e. relatively small changes in chemical structure can radically alter the activity of these drugs.
Let us finish with some important definitions. These are concepts which we will return to repeatedly throughout the course.
- Agonist is a drug which binds to its "receptor" and produces its characteristic effect. A drug may be a full agonist or partial agonist, depending on the maximal effect it produces. An antagonist binds to the receptor without causing an effect, thereby preventing an active substance from gaining access. Antagonists, like enzyme inhibitors, may be competitive, non-competitive or irreversible.
- Dose-Response. The sine qua non of drug effect. Simply put, as the dose of drug increases, the response should increase. [What if the response increases, then decreases as the dose is raised?] The curve generated is usually sigmoidal when effect is plotted against log dose (Dr. Strichartz will discuss the theoretical basis for this). Effect may be measured as a graded variable (change in blood pressure, force of contraction) or as a quantal variable (number dead/alive). The slope of the curve is characteristic of the particular drug-receptor interaction. When two drugs act by the same receptor mechanism, we expect to see two parallel log-dose response curves.
- ED50. The median effective dose, or the dose which produces a response in 50% of subjects. If the response is death (lethality) we call it the LD50. The EC50 refers to concentration rather than dose. Similar abbreviations are used for other response levels: ED99, LD1, etc.
- Potency. A terribly misused word – the lay public uses it to mean “effectiveness.” The potency of a drug refers to the dose (actually the molar concentration) required to produce a specific intensity of effect. [We usually specify the ED50, why?] If the ED50of drug A and B are 5 and 10 mg, respectively, the Relative Potency of A is twice that of B. Relative potency specifically applies to the comparison of drugs which act by the same mechanism, and therefore have parallel dose-response curves.
- Efficacy. Also called Maximal Efficacy or Intrinsic Activity. This is the maximum effect of which the drug is capable. A potent drug may have a low efficacy, and a highly efficacious drug may have a low potency. For the clinician, efficacy is much more important than potency (within limits). Who cares if the pill contains 5 or 10 mg of drug?
- Affinity. This refers to the strength of binding between drug and receptor. It is quantified by the dissociation constant kD (covered in the next lecture).
- Selectivity. This refers to the separation between desired and undesired effects of a drug. In the ideal case, a drug is completely specific, and an effective dose does not elicit any undesired effect. Penicillin is an example of a highly selective drug, since it works specifically by inhibiting cell wall synthesis, and (other than allergic responses) it has very little effect on human cells at normal doses. Unfortunately, many therapeutic agents, like digoxin and theophylline, produce dose-related side effects near their therapeutic dose range. For some drugs like cancer chemotherapeutic agents, their selectivity is their dose-limiting property, i.e., they are given to kill tumor cells until they produce toxicity in normal cells as well.
- Therapeutic Window. For every drug, there exists some concentration which is just barely effective (the Effective Concentration) and some dose which is just barely toxic (the Toxic Concentration). Between them is the therapeutic window where most safe and effective treatment will occur.
- Therapeutic Index. This is the ratio of toxic to effective doses at the level of 50% response: TD50/ED50. In animal toxicology studies, it is usually the LD50/ED50. Another measure sometimes utilized is the Certain Safety Factor, which is TD1/ED99.