1.8: Antiinflammatory Drugs
- Page ID
- 10629

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Inflammation is mediated in part by prostaglandins produced by the cyclooxygenase pathway. NSAIDs inhibit this pathway and serve as combined anti-inflammatory, anti pyretics, and analgesics. Because NSAIDs are generally nonspecific and exert numerous side effects, there is great interest in more specific therapeutics such as selective COX-2 inhibitors and anti-cytokine agents.
Prostaglandins: Physiologic and Pathologic Functions
All cells in the body have the capacity to synthesize prostaglandins. In response to inflammatory stimuli arachidonic acid (AA) is separated from plasma phospholipids by phospholipase A2. Cyclooxygenase metabolizes AA to the cycloendoperoxide prostaglandin H2 (PGH2), which is then converted to either PGD2, PGE2, PGF2α, PGI2(prostacyclin) or TXA2 (thromboxane) by appropriate enzymes (i.e. thromboxane synthase in platelets, prostacyclin synthase in endothelial cells).
The prostaglandins exert numerous physiologic and pathophysiologic functions :
- Physiologic: temperature homeostasis, bronchial tone, cytoprotection (gastric and renal mucosa), intestinal mobility, myometrial tone, semen viability (some prostaglandins like PGE1 have anti-inflammatory effects), renin secretion
- ƒ Pathologic: fever (aberrant hypothalamic thermoregulation), asthma (airway responsiveness and immune hyperreactivity), ulcers (loss of cytoprotection), diarrhea (intestinal mobility), dysmenorrhea (myometrial tone), inflammation, bone erosion, pain (thought to be caused by PGD2)
Specific functions of prostaglandins in the context of inflammation include:
- ƒ PGI2: inhibits platelet aggregation, vasodilatation, vascular permeability (edema)
- ƒ PGE2: pain, hyperalgesia, heat, vasodilatation, bronchoconstriction, synergistically act with other pro-inflammatory mediators (histamine, complement, LTB4)
- ƒ TXA2: promotes platelet aggregation, vasoconstriction, bronchoconstriction
Cyclooxygenase
There are two forms of cyclooxygenase (COX) enzymes: COX-1 and COX-2. Though COX-1 and COX-2 catalyze the same reaction, their expression, functions, and properties are markedly different.
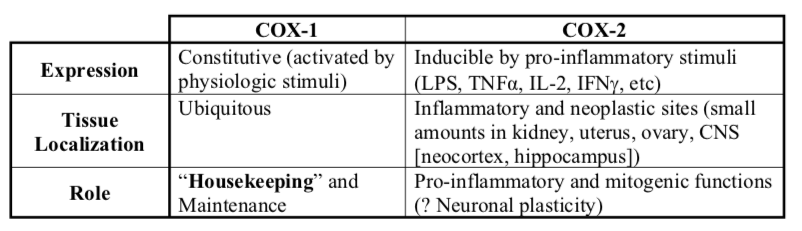
COX-1 produces PGE2, PGI2, and TXA2 in platelets, GI mucosa, vascular endothelium, and the kidney. The housekeeping functions of these prostaglandins include maintaining renal and gastrointestinal blood flow (cytoprotection), regulation of vascular homeostasis, renal function, intestinal mucosal proliferation, and platelet function.
Pro-inflammatory functions of COX-2 produced prostaglandins include pain, fever, leukocyte proliferation, and inflammation. COX-2 produces prostaglandins at sites of inflammation (in macrophages, in synovial tissue of rheumatoid arthritis joint). Mitogenic functions of COX-2 produced prostaglandin include renal genesis and reproduction.
The goal of pharmacologic anti-inflammatory therapy has been to inhibit COX-2 produced prostaglandins. Non-specific inhibition of COX-1 results in gastrointestinal and platelet side effects. Recent data on the toxicity of COX-2 selective nsaids illustrate that this is an overly simplistic view. The magnitude of the COX-2 problem is still unclear at this writing, but it will be considered at various points in this discussion.
There is an entire additional pathway of arachidonic acid metabolism by enzymes called lipoxygenases. 5-lipoxygenase is not present in all tissues but is limited to neutrophils, eosinophils, monocytes, and certain mast cell populations. Lipoxygenases produce leukotrienes (e.g. LTB4, LTD4), which are potent bronchoconstrictors and chemotactic agents. Leukotrienes have important roles in asthma, glomerulonephritis and inflammatory bowel disease. (Refer to the Asthma case.
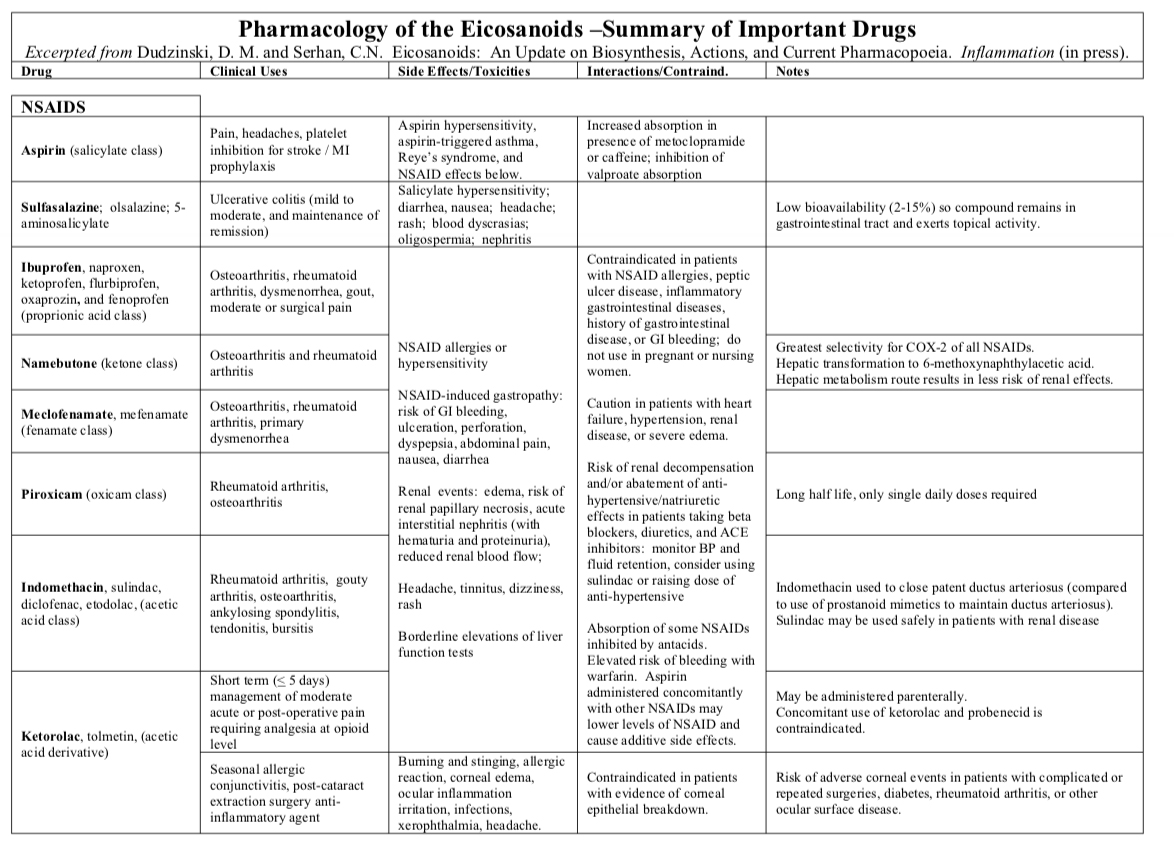
NSAIDs (Non-steroidal anti-inflammatory drugs)
Most NSAIDs are polycyclic carboxylic acid derivatives with relatively low pKa values. NSAIDs are often classified on the basis of their chemical structure (see Figure 1).
- ƒ Salicylates: aspirin; diflunisal, 5-aminosalicylate, sodium salicylate, magnesium salicylate, sulfasalazine, olasalzine
- ƒ Acetic acids: indomethacin, diclofenac, sulindac, etodolac, ketorolac, tolmetin
- ƒ Propionic acids: ibuprofen, naproxen, fenoprofen, ketoprofen, flurbiprofen, oxaprozin
- ƒ Fenamic acids: meclofenamate, mefenamate
- ƒ Enolic acids (oxicam class): piroxicam
- ƒ Ketones: nabumetone (converted to 6-naphthylacetic acid in liver)
NSAID General Pharmacodynamics
All NSAIDs (except aspirin) act as reversible, competitive cyclooxygenase inhibitors. They block the hydrophobic channel by which the substrate arachidonic acid accesses the enzyme active site. Aspirin covalently modifies and destroys the cyclooxygenase enzyme.
The ultimate function of the NSAID is to inhibit COX-2, preventing generation of proinflammatory eicosanoids, and thus limiting the extent of inflammation and adverse signs and symptoms.
- ƒ All NSAIDs have a ratio of inhibition of COX-2 / inhibition of COX-1. The higher the ratio, the more specific the therapeutic effect and fewer GI or platelet effects.
− NSAIDs with high ratio (100:1 to 1000:1) are COX-2 Selective (Coxib)ƒ
- Despite the benefits of NSAIDs, they only provide symptomatic relief, as the underlying pathophysiology or injury generally is unaffected.
NSAIDs have three primary therapeutic effects:
- ƒ Analgesia
- ƒ Anti-pyrexia (decreasing hypothalamic PGE2)ƒ
- Anti-inflammatory
NSAIDs are also used as anti-thrombotics. Since they impair platelet aggregation, they prolong bleeding time, and function as anticoagulants. The COX-2 specific inhibitors do not exert anti-thrombotic effects. Other functions of NSAIDs include inhibition of:
- Superoxide generation
- ƒ Lysosomal enzyme release
- ƒ Neutrophil aggregation / adhesion
- Lymphocyte function
- ƒ Cytokine release (IL-6)
Indications for Specific NSAIDs
Please refer to Table 1 to find indications common to each structural class of NSAID. See below for COX-2 selective drugs.
Non-selective NSAIDs are used as analgesics for moderate pain of musculoskeletal and inflammatory origin (headaches, dysmenorrhea, osteoarthritis, rheumatoid arthritis, gout, surgical pain, tendonitis, and bursitis). NSAIDs also function as anti-inflammatory agents in many of these conditions and ulcerative colitis. Aspirin anti-platelet effects are used for MI and stroke prophylaxis. Acetaminophen (technically not an NSAID) has no anti-inflammatory activity but is widely used as an analgesic and anti-pyretic.
NSAID Pharmacokinetics
As stated previously, NSAIDs are weak organic acids. They generally haveƒ
- Efficient GI absorption (nearly complete)
- ƒLow first pass hepatic metabolism
- Small volumes of distribution but extensive protein binding (>95%) which slows the rate at which these drugs cross the capillary wall and penetrate tissue.
- ƒ Accumulation in cells at sites of inflammation (acidic NSAIDs are preferentially sequestered in inflamed synovial tissues)
- ƒ Efficient enterohepatic and renal excretion
- ƒ Variable half lives (the lower the pKa generally the shorter the half-life)
Plasma Elimination Half Lives: Another method to classify NSAIDs (besides structure)
- Short Half Life (< 6 hours): more rapid effect and clearance Aspirin (0.25-0.33 hrs), Diclofenac (1.1 ± 0.2 hrs), Ketoprofen (1.8 ± 0.4 hrs), Ibuprofen (2.1 ± 0.3 hrs), Indomethacin (4.6 ± 0.7 hrs)
- ƒ Long Half Life (> 10 hours): slower onset of effect and slower clearance Naproxen (14 ± 2 hrs), Sulindac (14 ± 8hrs), Namebutone (26 ± 5 hrs), Piroxicam (57 ± 22 hrs) (also COX-2 Selective Inhibitors)
Important Drug Interactions:
- Displace other drugs from plasma protein binding sites:
- Anti-coagulants (warfarin): Bleeding risk greatly increased
- Phenytoin: (increased CNS toxicity, difficulty dosing)
- Oral Hypoglycemics: (increased hypoglycemic risk)
- Methotrexate: (increased toxicity)
- Anti-Hypertensives (diuretics, beta blockers, ACE inhibitors): NSAIDs may blunt the anti-hypertensive effects and cause renal decompensation or renal failure in patients receiving these drugs
- Methotrexate, digoxin, aminoglycosides, lithium: NSAIDs inhibit clearanceƒ
- Probenecid: renal clearance of NSAIDs reduced by probenecid
- ƒ Antacids: absorption of some NSAIDs inhibited by antacids
- ƒ Aspirin: may lower levels of other NSAIDs, but side effects are additive
NSAID Toxicity
NSAIDs affect the gastrointestinal, CNS, hepatic, renal, hematologic, and skin systems. NSAIDs also cause allergic phenomena.
Gastrointestinal Toxicity of NSAIDs
Prostaglandins suppress gastric acid secretion and help maintain gastric mucosal barrier, thus providing gastrointestinal protection. Because of their suppression of prostaglandin synthesis NSAIDs tend to cause gastric irritation, exacerbate peptic ulcer disease, cause mucosal lesions (superficial to penetrating ulcers), and may induce bleeding.
NSAID induced gastropathy typically includes gastritis, gastric bleeding, mucosal and subepithelial damage, and erosions, which may progress to ulcerations and perforations.
- ƒ Occult blood loss may occur and massive GI bleeding may also develop.
- ƒ Symptoms including pain, dyspepsia, nausea, vomiting are frequent
- ƒ Overall, there is poor correlation of these symptoms with endoscopic findings.ƒ
- NSAID induced gastric toxicity causes great morbidity, requiring annual care expenditures of $4 billion, and causes 7500 deaths per year.
- FDA estimates that ulcers, bleeding or perforation occur in 1 to 2 % of patients using NSAIDs for three months and 2 to 5% of those using them for one year.
- ƒSpecific risk factors for NSAID induced GI toxicity include: higher NSAID doses, older age, concurrent steroid use, history of peptic ulcer disease
Treatment of NSAID Induced GI Toxicity
- ƒ Discontinuation / Avoidance of NSAIDs / Use “Gastroprotective” NSAIDsƒ
- Take medication with meal
- ƒPharmacologic
- H2 Receptor Antagonists (high doses of ranitidine)
- Proton Pump Inhibitors (omeprazole)
- Misoprostol (PGE1 analog which restores cytoprotective effects)
- Sucralfate
- COX-2 Specific NSAIDs – use now called into question (see discussion below)
- Reduce risks of ulceration, bleeding, perforation vs. nonselective NSAIDs
CNS Toxicity:
CNS toxicity includes headache, confusion, tinnitus (aspirin), dizziness, mood alteration and depression, and aseptic meningitis (particularly in SLE patients). Aspirin is linked toReye’s Syndrome (below).
Hepatic Toxicity:
NSAIDs may cause asymptomatic elevations of liver enzyme, or transaminitis (most common with diclofenac. Acute idiosyncratic hepatitis has also been reported. Reye’s Syndrome is an often fatal combination of microvesicular steatosis and hepatic encephalopathy thought to be caused by the administration of aspirin to children post febrile viral infection (VZV, influenza B). For this reason, aspirin is generally not given to children.
Nephrotoxicity:
In healthy individuals with normal kidneys PGE2 and PGI2 play no role in controlling renal function. Under certain conditions of localized circulatory stress often associated
with elevated levels of angiotensin II and catecholamines, locally produced vasodilating prostaglandins become essential to the maintenance of adequate renal function.
Inhibition of these vasodilating prostaglandins decreases renal renal blood flow and GFR and may cause tissue injury. Patients at most risk include those with congestive heart failure, volume depletion, chronic renal disease, liver disease and those patients receiving diuretics. Nephrotoxic effects of NSAIDs include edema, high blood pressure, increased creatinine, and hyperkalemia. Hypertension and edema have been seen with both selective and non-selective NSAIDs. NSAIDs may ultimately cause renal ischemia or failure, nephrotic syndrome, interstitial nephritis (most commonly with fenoprofen), renal papillary necrosis, and calculi.
Hematologic Effects:
An effect on platelet aggregation persists for as long as the NSAIDs are present. They should be discontinued for a long enough period before surgery to permit complete excretion (i.e., 4 to 5 times the half-life). Aspirin should be discontinued 7-10 days prior in order to give sufficient time to make new platelets. NSAIDs can interfere with the therapeutic antiplatelet effect of aspirin if the drugs are taken together.
Blood dyscrasias such as agranulocytosis, thrombocytopenia and aplastic anemia are rarely associated with NSAIDs.
Cutaneous and Hypersensitivity Effects:
NSAIDs can cause urticaria, bronchospasm, anaphylaxis, and erythema multiforme. A wide variety of skin reactions most frequently reported with piroxicam and benoxaprofen (withdrawn from market) include photosensitivity reactions, exfoliative dermatitis, Stevens-Johnson syndrome, and toxic epidermal necrolysis.
NSAID-induced hyperreactivity: In patients with aspirin allergy, NSAID exposure is more likely to cause ocular and nasal congestion, severe bronchospasm, and possible anaphylactic reaction. Possible etiology is shunting of arachadonic acid to lipoxygenase pathway leading to increased synthesis of bronchoconstrictor leukotrienes.
- ƒ Samter's triad: aspirin allergy / hypersensitivity higher in patients with nasal polyps, bronchial asthma, and rhinitis (sinusitis).
- ƒ Occurs in 10% of asthmatics.
Other Toxicities Unique to Aspirin:
Aspirin overdose can cause metabolic acidosis but also stimulates the medullary respiratory center, causing respiratory alkalosis.
Salicylism refers to a syndrome of chronic, excessive aspirin dosing characterized by nausea, vomiting, diarrhea, and dehydration, hyperventilation, headache, tinnitus, visual and auditory disturbances, confusion, stupor, and delirium.
COX-2 Selective Inhibitors: The Coxibs
The coxibs represent a subset of NSAIDs that preferentially block the hydrophobic substrate channel in COX-2. Currently approved coxibs include celecoxib and valdecoxib. These drugs are approved for rheumatoid arthritis, osteoarthritis, pain, primary dysmenorrhea, and familial adenomatous polyposis (they decrease the number and size of adenomas in patients with history of FAP).
- The potential therapeutic role of coxibs in Alzheimer’s disease is being studied (COX-2 is the predominant isoform in the neocortex and hippocampus).
- ƒ COX-2 is induced by LH prior to ovulation and at delivery. COX-2 selective inhibitors may have a role in preventing preterm labor and delivery.
Effects and Toxicities of COX-2 Selective Drugs:
Rofecoxib was recently withdrawn from the market when an increased rate of myocardial infarction and stroke was seen in a placebo-controlled trial for FAP. There is now concern that all COX-2 inhibitors may increase the risk of thrombotic events during chronic therapy. Evidence for this has now appeared in one trial of valdecoxib and one study of celecoxib. A possible explanation may be that coxibs can inhibit endothelial prostaglandin synthesis but lack a compensatory effect on platelet thromboxane synthesis. The situation is currently unsettled, but it seems prudent to restrict the use of these drugs to patients for whom the potential benefits are clearly worth the risk. The risk vs. benefit may be difficult to characterize: COX-2 inhibitors were designed in part to limit the gastrotoxicity associated with NSAIDs. Although the rate is lower, events still occur and symptoms are similar. However, events are less serious (i.e. less perforation) vs. conventional NSAIDs.
Platelets only express COX-1, so COX-2 has no effect on platelet function or the production of TXA2. The implications of this are:
- ƒPatients on MI prophylaxis still need aspirin even if they are on a COX-2 selective inhibitor
- ƒCoxibs unlike NSAIDs may be administered safely with warfarin.
COX-2 is present in the kidney and knock out mice show kidney inflammation and papillary changes. This suggests that chronic treatment with coxibs may impair normal renal development and function. Some evidence suggests that inhibition of COX-2 may generate problems with wound healing and angiogenesis.
Glucocorticoids
Glucocorticoids are 21-carbon steroid molecules with a variety of physiologic and metabolic effects. Cortisol (hydrocortisone) is the principal circulating glucocorticoid in humans. Glucocorticoid activity depends on presence of a hydroxyl group at carbon number 11 in the steroid molecule. Cortisone and prednisone lack glucocorticoid activity until converted to cortisol and prednisolone in the liver (by reducing the C=O at carbon 11 to a hydroxyl). All glucocorticoid preparations marketed for topical use are 11 beta hydroxyl compounds, thus eliminating the need for hepatic transformation.
Commonly used glucocorticoids are characterized short, medium, and long acting on the basis of ACTH suppression after a single dose (of equivalent anti-inflammatory activity to 50 mg of prednisone). The relative potency of the glucocorticoids correlates with their affinity for the glucocorticoid receptor. The observed potency of a glucocorticoid is a measure not only of the intrinsic biological potency but also the duration of action. However, relationships between the circulating half-life and duration of action, and between circulating half-life and glucocorticoid potency are imprecise.
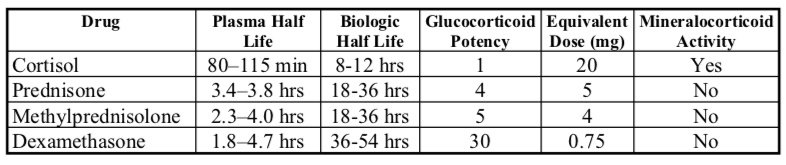
It has been suggested that the duration of action of a glucocorticoid is not determined by its presence in the circulation. Steroids pass through the cell membrane and enter the cytoplasm where they bind to a specific cytoplasmic receptor protein. These glucocorticoid receptors belong to a superfamily of DNA binding proteins that affect gene regulation. Glucocorticoids alter transcriptional regulation of specific cytokine genes. Therefore, the effects of glucocorticoids continue to act within the cell after glucocorticoids have disappeared from the circulation (note disparities in plasma and biologic half lives).
Therapeutic Effects of Glucocorticoids
Glucocorticoids are anti-inflammatory and immunosuppressive agents. Glucocorticoids administered at pharmacologic doses inhibit the action of COX-2 by decreasing the expression of COX-2 and the cytokines that activate it. They limit the available pool of arachidonic acid substrate by inhibiting phospholipase A2 (via the lipocortin pathway). These combined actions create a powerful anti-inflammatory effect because virtually all eicosanoid pathways are inhibited. Because of this profound and global suppression, glucocorticoids are indicated for a number of autoimmune and inflammatory conditions.
Effects of Glucocorticoids on Humoral Factors
- ƒMild decrease in immunoglobulin levels
- ƒDecreased RE clearance
- ƒDecreased synthesis of prostaglandins and leukotrienes

Glucocorticoid Toxicities
Glucocorticoids exhibit a diverse array of toxicities (see Table). Recall that a condition of glucocorticoid excess is a Cushingoid syndrome.

Steroid Withdrawal and Glucocorticoid Replacement
Because of suppression of the pituitary-adrenal axis by chronic glucocorticoid therapy, patients that undergo surgical procedures or acute medical illness should receive stress dose steroids generally equivalent to 300 mg of hydrocortisone administered as a split dose over a 24 hour period. Patients may require very slow and low reductions of steroids back to baseline to minimize the symptoms of steroid withdrawal, which include joint and muscle pain, nausea, lethargy, weight loss, and fever.
Anti-Cytokine Agents
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune and inflammatory disease that primarily attacks the joints. Autoimmune targeting of normal joint proteins results in inflammation with local release of cytokines, especially TNFα, growth factors, and interleukins, all of which induce COX-2 expression. Levels of TNFα, COX-2, and PGE2are markedly elevated in the synovial fluid of affected joints. PGE2 binds to synovial cell receptors on and stimulates release of matrix metalloproteinases (MMPs) which directly damage joint tissue. TNFα also stimulates production of IL-1 and IL-6; these pro- inflammatory mediators along with COX-2 derived prostaglandins activate the surrounding endothelial tissue to recruit inflammatory cells.
The newest therapeutic agents inhibit the pro-inflammatory effects of the cytokines TNFα and IL-1. By limiting TNF activity, the generation of pro-inflammatory cytokines IL-1 and IL-6 is diminished. Three strategies have emerged: 1) creating monoclonal antibodies to the TNFα and IL-1 proteins, 2) solubilizing forms of the endogenous receptors of TNFα, and 3) making recombinant versions of endogenous receptor antagonist.
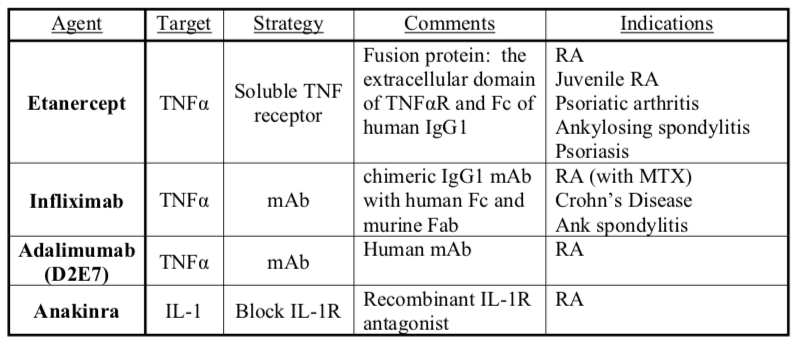
Comparison of Infliximab, Etanercept, and Adalimumab in rheumatoid arthritis:
- ƒInfliximab administered IV (by doctor); etanercept and adalimumab given subcutaneously (by patient)
- ƒInfliximab 3 mg/kg loading dose with similar doses at 2 weeks, 6 weeks, and then every 8 weeks thereafter. Etanercept 25 mg twice weekly or 50 mg weekly. Adalimumab 40 mg every other week.
- ƒInfliximab must be administered in combination with methotrexate. Etanercept and adalimumab may be monotherapy or may be combined with methotrexate.
- ƒAll reduce signs, symptoms, and structural damage (joint erosion on radiograph).
Anti-Cytokine Agent Toxicities:
The anti-cytokine agents blunt the immune response, and thus cause many side effects that result from immunosuppression (infections, and loss of tumor surveillance). The body’s response to the foreign protein also poses risk of development of antibodies.
- Injection site reactions
- ƒHypersensitivity reactions (i.e. to murine protein)ƒ
- Opportunistic Infections
- Tuberculosis
- Fungi (Aspergillus)
- Pneumocystiis carinii
- Listeria
- Bacterial Sepsis
- Lymphoproliferative Disorders
- Lupus like Syndrome
- Autoantibodies and Antibodies to drug
- Rare aplastic anemia, demyelinating syndrome
Future Applications of Anti-Cytokine Therapies
Because of the broad role of TNFα in disease, there is interest in applying these drugs to conditions as varied as vasculitis, myositis, GVHD, uveitis, CHF, sarcoidosis, psoriasis, ARDS, Still’s disease, Wegener’s syndrome, etc.